Wang Hai-Yan, Qi Meng, Sun Ming-Fei, Li Dong-Fang, Wang Rong-Jun, Zhang Su-Mei, Zhao Jin-Feng, Li Jun-Qiang, Cui Zhao-Hui, Chen Yuan-Cai, Jian Fu-Chun, Xiang Rui-Ping, Ning Chang-Shen, Zhang Long-Xian
Experimental and Research Center, Henan University of Animal Husbandry and Economy, Zhengzhou, China.
College of Animal Science and Veterinary Medicine, Henan Agricultural University, Zhengzhou, China.
Front Microbiol. 2019 Jun 25;10:1399. doi: 10.3389/fmicb.2019.01399. eCollection 2019.
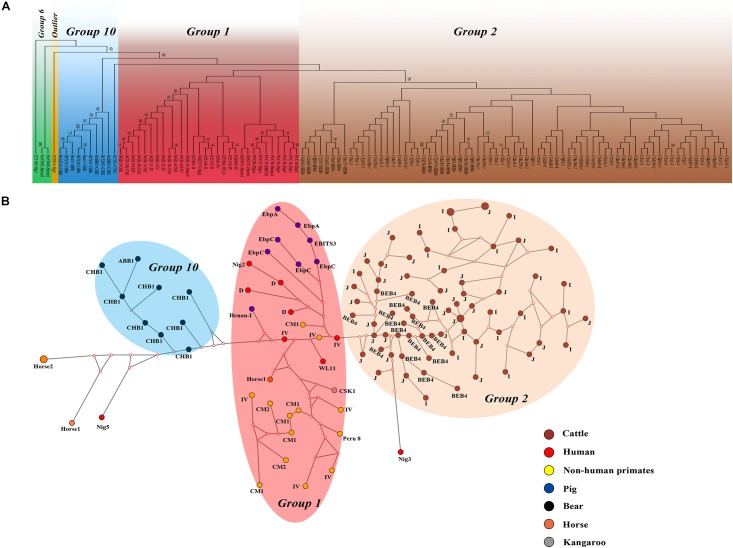
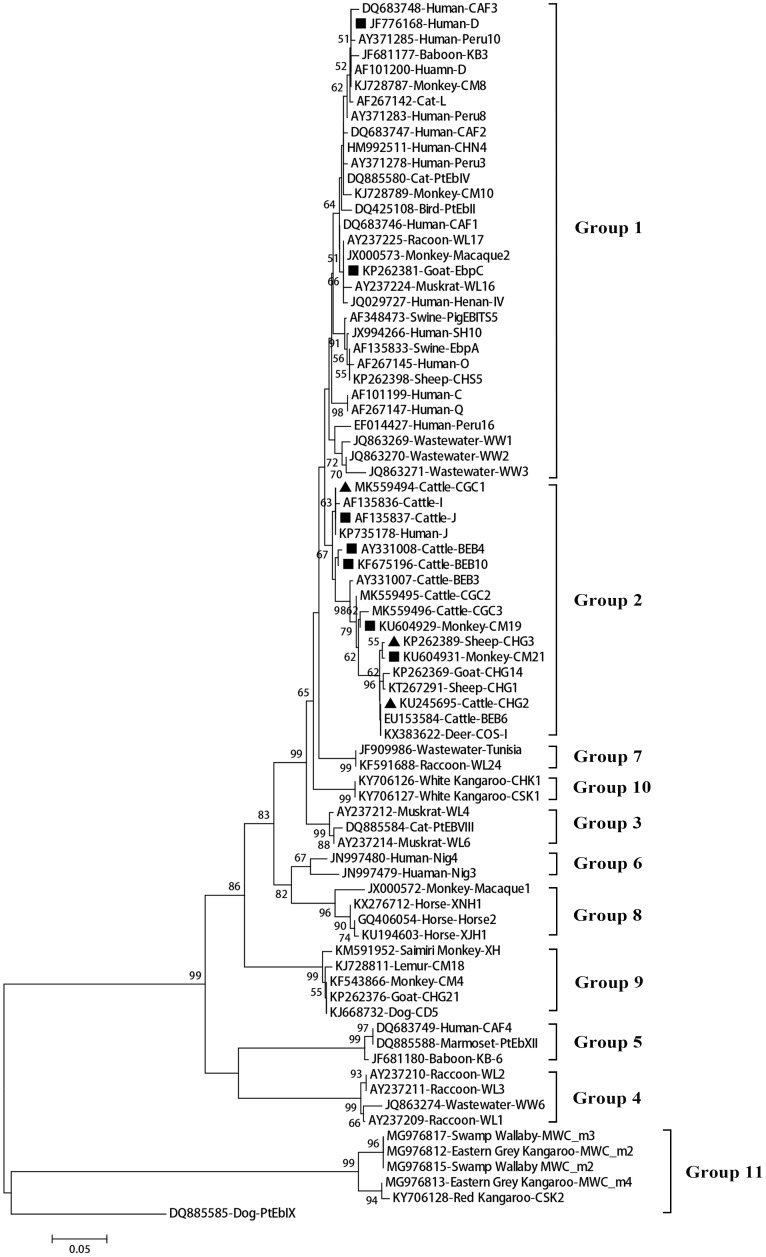
, an obligate intracellular pathogen, can infect various hosts. In this study, 3527 dairy cattle fecal specimens were collected from different geographic locations in China (including 673 from Shandong province, 1,440 from Guangdong province and 1,414 from Gansu province) and examined for the presence of using polymerase chain reactions targeting the ribosomal internal transcribed spacer (ITS). The dominant genotypes identified were further subtyped by multilocus sequence typing. The overall prevalence of was 14.2% (501/3527), with a significant difference in prevalence among the different geographical locations ( < 0.001). Our logistic regression analysis showed that all four variables (farming model, location, age, and clinical manifestations) had strong effects on the risk of contracting . Sequence analysis revealed 11 genotypes: eight known genotypes (J, I, BEB4, BEB10, D, EbpC, CM19, and CM21) and three novel genotypes (named here as CGC1, CGC2, and CGC3). Genotypes J and I, the commonest, were found on all farms across the three provinces. Our linkage disequilibrium analysis showed a clonal population structure in the dairy cattle population but the ITS genotypes had different population structures. Phylogenetic and haplotype network analysis showed the absence of geographical segregation in the dairy cattle populations. Instead, they revealed the presence of host adaptation to the populations in various animals. Our findings augment the current understanding of transmission dynamics.
作为一种专性胞内病原体,可感染多种宿主。在本研究中,从中国不同地理位置收集了3527份奶牛粪便标本(包括山东省的673份、广东省的1440份和甘肃省的1414份),并使用针对核糖体内部转录间隔区(ITS)的聚合酶链反应检测是否存在[病原体名称未给出]。通过多位点序列分型对鉴定出的优势基因型进一步进行亚型分类。[病原体名称未给出]的总体患病率为14.2%(501/3527),不同地理位置的患病率存在显著差异(P<0.001)。我们的逻辑回归分析表明,所有四个变量(养殖模式、地点、年龄和临床表现)对感染[病原体名称未给出]的风险都有强烈影响。序列分析揭示了11种基因型:8种已知基因型(J、I、BEB4、BEB10、D、EbpC、CM19和CM21)和3种新基因型(此处命名为CGC1、CGC2和CGC3)。最常见的基因型J和I在三省的所有养殖场都有发现。我们的连锁不平衡分析表明,[病原体名称未给出]奶牛群体中存在克隆群体结构,但ITS基因型具有不同的群体结构。系统发育和单倍型网络分析表明,[病原体名称未给出]奶牛群体中不存在地理隔离。相反,它们揭示了在各种动物中宿主对[病原体名称未给出]群体的适应性存在。我们的研究结果增强了对[病原体名称未给出]传播动态的当前理解。