Abarca Nadia, Santín Mónica, Ortega Sheila, Maloney Jenny G, George Nadja S, Molokin Aleksey, Cardona Guillermo A, Dashti Alejandro, Köster Pamela C, Bailo Begoña, Hernández-de-Mingo Marta, Muadica Aly S, Calero-Bernal Rafael, Carmena David, González-Barrio David
Parasitology Reference and Research Laboratory, National Centre for Microbiology, Majadahonda, 28220 Madrid, Spain.
Department of Veterinary Sciences, Biomedical Sciences Institute, Autonomous University of Ciudad Juárez, Chihuahua 32310, Mexico.
Vet Sci. 2021 Sep 11;8(9):191. doi: 10.3390/vetsci8090191.
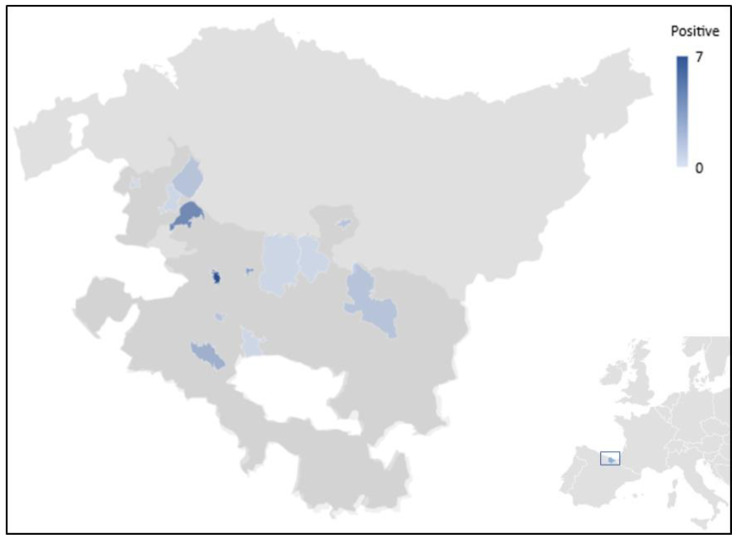
Some enteric parasites causing zoonotic diseases in livestock have been poorly studied or even neglected. This is the case in stramenopile sp. and the microsporidia in Spain. This transversal molecular epidemiological survey aims to estimate the prevalence and molecular diversity of sp. and in cattle faecal samples ( = 336) in the province of Álava, Northern Spain. Initial detection of and was carried out by polymerase chain reaction (PCR) and Sanger sequencing of the small subunit () rRNA gene and internal transcribed spacer (ITS) region, respectively. Intra-host subtype diversity was further investigated by next generation amplicon sequencing (NGS) of the rRNA gene in those samples that tested positive by conventional PCR. Amplicons compatible with sp. and were observed in 32.1% (108/336, 95% CI: 27.2-37.4%) and 0.6% (2/336, 95% CI: 0.0-1.4%) of the cattle faecal samples examined, respectively. Sanger sequencing produced ambiguous/unreadable sequence data for most of the isolates sequenced. NGS allowed the identification of 10 subtypes including ST1, ST3, ST5, ST10, ST14, ST21, ST23, ST24, ST25, and ST26. All -positive isolates involved mixed infections of 2-8 STs in a total of 31 different combinations. The two sequences were confirmed as potentially zoonotic genotype BEB4. Our data demonstrate that mixed subtype infections are extremely frequent in cattle in the study area. NGS was particularly suited to discern underrepresented subtypes or mixed subtype infections that were undetectable or unreadable by Sanger sequencing. The presence of zoonotic ST1, ST3, and ST5, and BEB4 suggest cross-species transmission and a potential risk of human infection/colonization.
一些可在牲畜中引发人畜共患病的肠道寄生虫尚未得到充分研究,甚至被忽视。西班牙的卵菌纲物种和微孢子虫便是如此。这项横向分子流行病学调查旨在估计西班牙北部阿拉瓦省牛粪便样本(n = 336)中卵菌纲物种和微孢子虫的流行率及分子多样性。分别通过聚合酶链反应(PCR)以及小亚基(SSU)rRNA基因和内转录间隔区(ITS)区域的桑格测序对卵菌纲物种和微孢子虫进行初步检测。对于那些通过常规PCR检测呈阳性的样本,通过下一代扩增子测序(NGS)对rRNA基因进一步研究宿主内卵菌纲物种亚型多样性。在所检测的牛粪便样本中,分别有32.1%(108/336,95%置信区间:27.2 - 37.4%)和0.6%(2/336,95%置信区间:0.0 - 1.4%)观察到与卵菌纲物种和微孢子虫相符的扩增子。对于大多数测序的卵菌纲物种分离株,桑格测序产生了模糊/不可读的序列数据。NGS鉴定出10种卵菌纲物种亚型,包括ST1、ST3、ST5、ST10、ST14、ST21、ST23、ST24、ST25和ST26。所有卵菌纲物种阳性分离株均涉及2 - 8种亚型的混合感染,共有31种不同组合。这两个微孢子虫序列被确认为潜在的人畜共患病基因型BEB4。我们的数据表明,在研究区域的牛中,卵菌纲物种混合亚型感染极为常见。NGS特别适合识别桑格测序无法检测到或不可读的代表性不足的亚型或混合亚型感染。人畜共患病卵菌纲物种亚型ST1、ST3和ST5以及微孢子虫BEB4的存在表明存在跨物种传播以及人类感染/定植的潜在风险。