Department of Medical Research, Chi-Mei Medical Center, Tainan, Taiwan.
Cardiovascular Institute, An Nan Hospital, China Medical University, Tainan, Taiwan.
J Cell Mol Med. 2019 Sep;23(9):6378-6392. doi: 10.1111/jcmm.14526. Epub 2019 Jul 17.
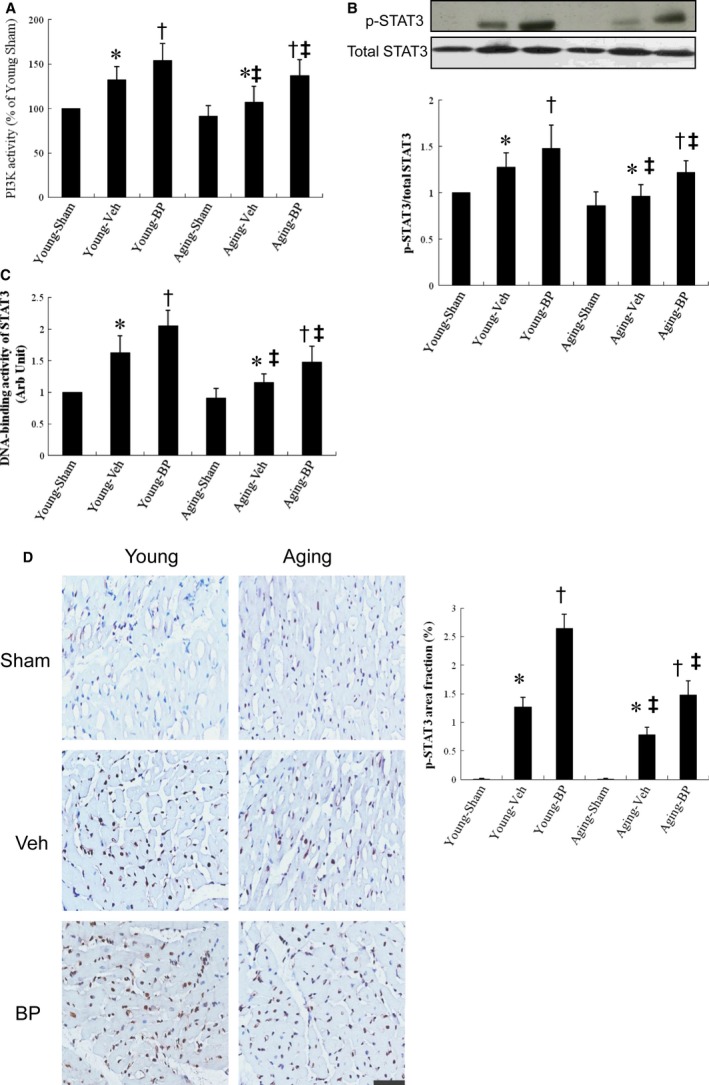
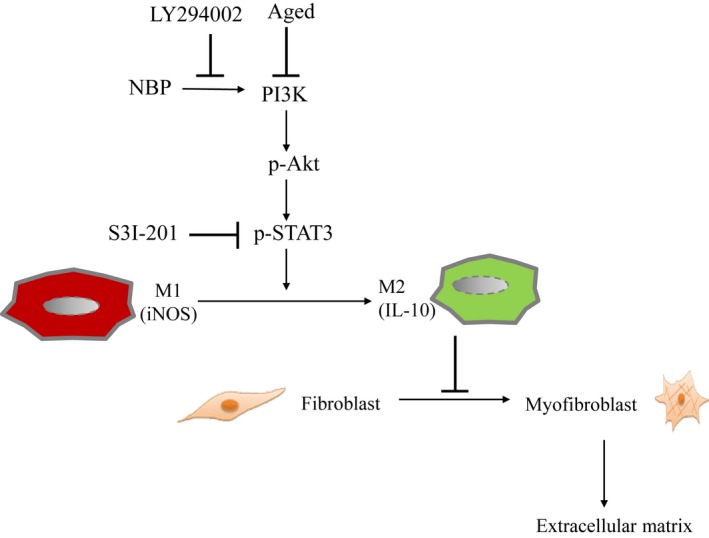
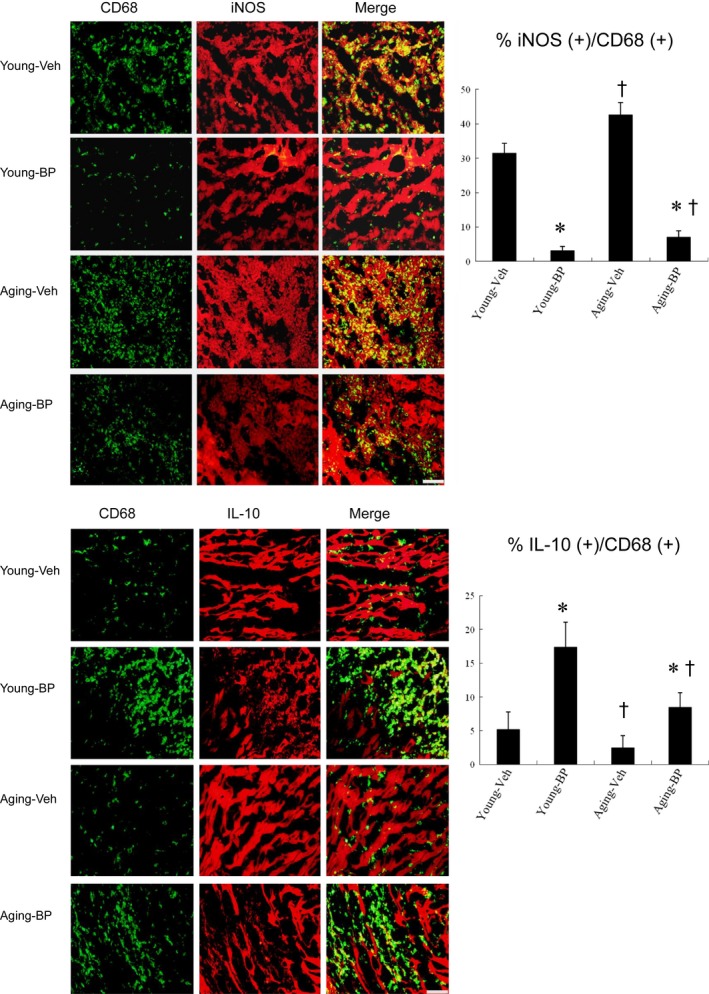
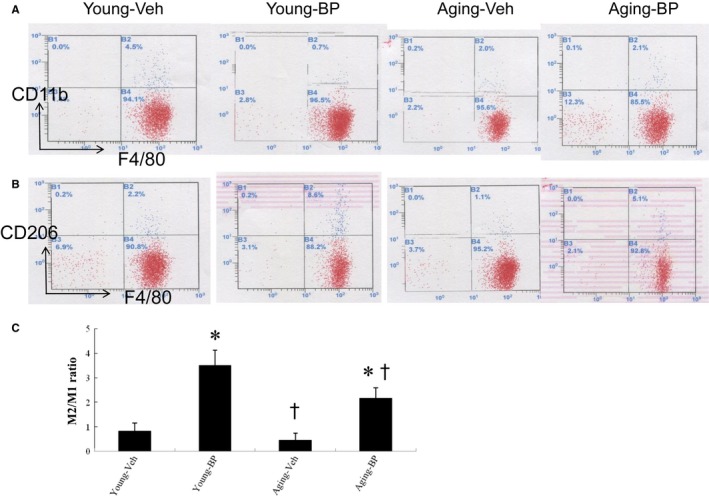
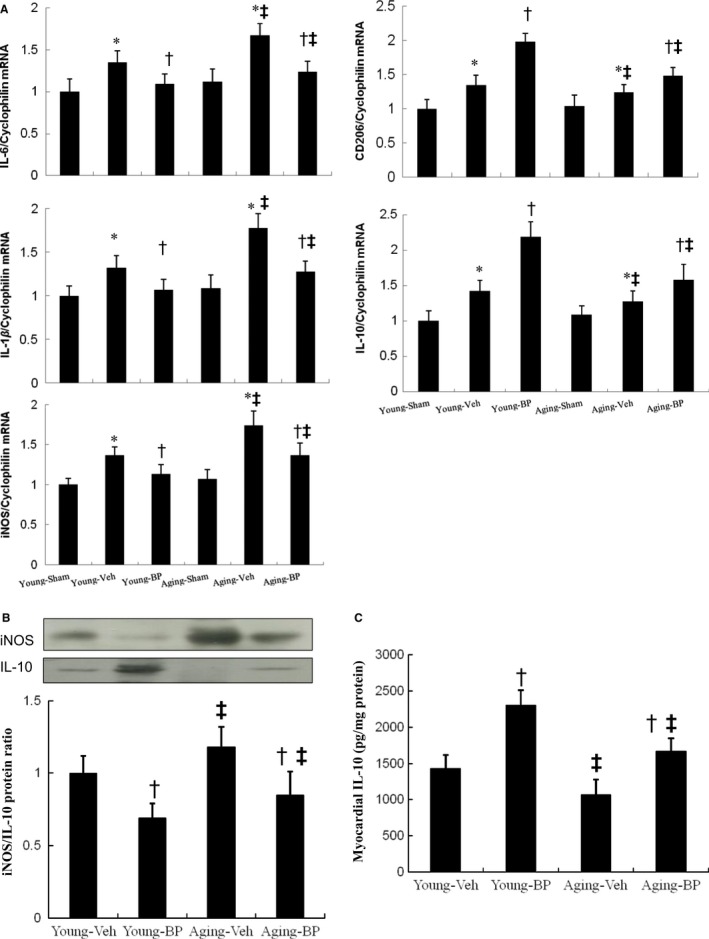
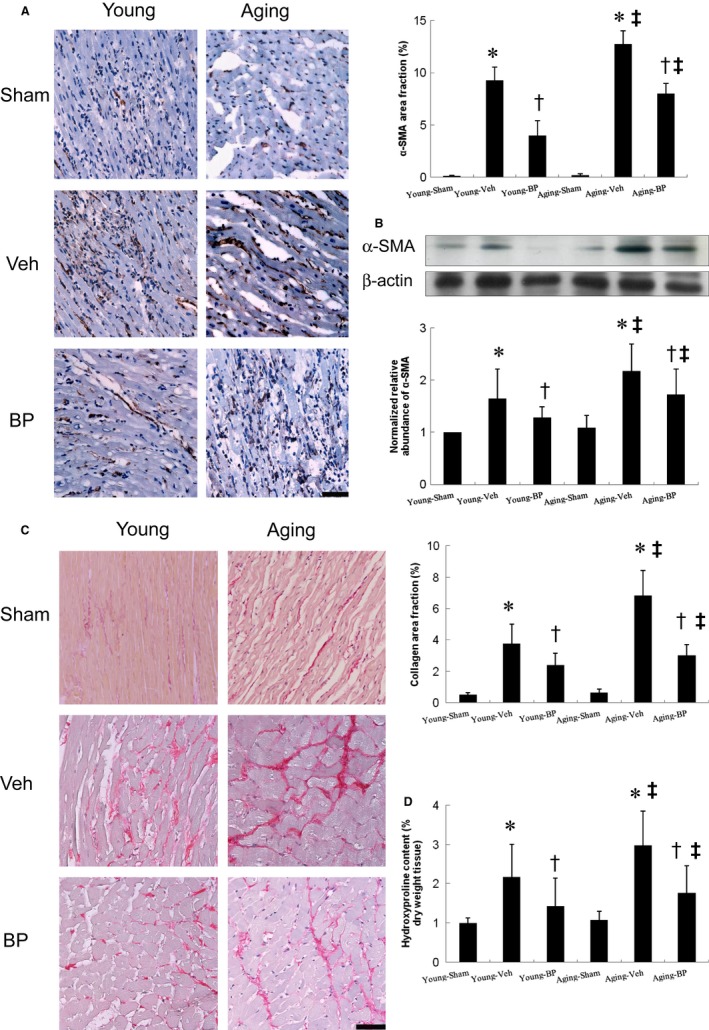
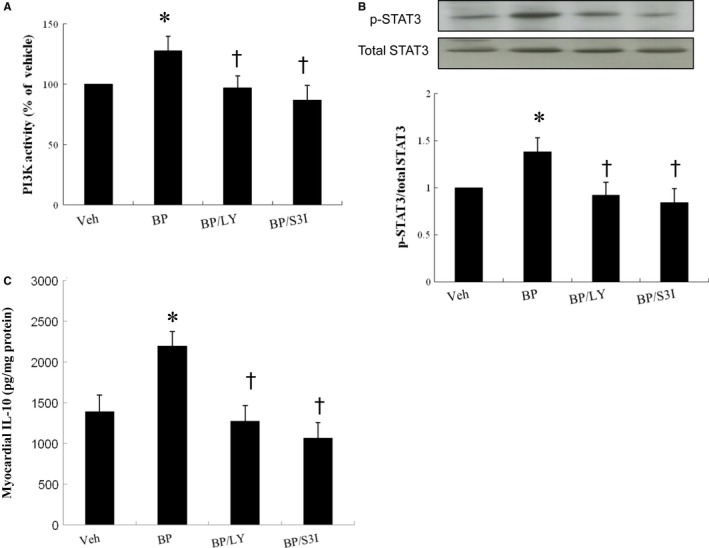
Ageing is associated with impaired repair mechanisms in cardiovascular diseases. Macrophages contribute to cardiac fibrosis after myocardial infarction (MI). The phosphatidyl-inositol-3-kinase (PI3K) pathway has been shown to play a role in cardiac remodelling after MI. It remained unclear whether n-butylidenephthalide, a major component of Angelica sinensis, can attenuate cardiac fibrosis by regulating the PI3K/signal transducer and activator of transcription 3 (STAT3)-mediated macrophage phenotypes in ageing rats after MI. Twenty-four hours after ligation of the left anterior descending artery, young (2-month-old) and ageing (18-month-old) male Wistar rats were treated with either vehicle or n-butylidenephthalide for 4 weeks. There were similar infarct sizes in both age groups. Compared with young rats, ageing rats exhibited significant increased cardiac fibrosis after MI, which can be attenuated after administering n-butylidenephthalide. MI was associated with decreased activities of PI3K and STAT3 in ageing rats compared with young rats. In both age groups, n-butylidenephthalide effectively provided a significant increase of STAT3 phosphorylation, STAT3 activity, STAT3 nuclear translocation, myocardial IL-10 levels and the percentage of M2c macrophage and a decrease of myofibroblast infiltration. The effects of n-butylidenephthalide on increased IL-10 levels were reversed by LY294002 or S3I-201. Furthermore, LY294002 abolished the STAT3 phosphorylation, whereas PI3K activity was not affected following the inhibition of STAT3. In conclusions, the host environment is responsible for ageing-related myofibroblast dysregulation in response to MI which can be improved by administering n-butylidenephthalide via macrophage differentiation towards M2 phenotype by targeting the PI3K/STAT3 axis.
衰老是心血管疾病中受损修复机制的相关因素。巨噬细胞在心肌梗死(MI)后导致心肌纤维化。已经表明,磷脂酰肌醇-3-激酶(PI3K)途径在 MI 后心脏重塑中发挥作用。当归的主要成分丁烯基苯酞是否可以通过调节 PI3K/信号转导和转录激活因子 3(STAT3)介导的 MI 后衰老大鼠巨噬细胞表型来减轻心脏纤维化,这一点尚不清楚。在左前降支结扎 24 小时后,年轻(2 个月大)和衰老(18 个月大)雄性 Wistar 大鼠分别用载体或丁烯基苯酞治疗 4 周。两组的梗死面积相似。与年轻大鼠相比,衰老大鼠在 MI 后表现出明显增加的心脏纤维化,给予丁烯基苯酞后可以减轻。与年轻大鼠相比,MI 导致衰老大鼠的 PI3K 和 STAT3 活性降低。在两个年龄组中,丁烯基苯酞均可有效显著增加 STAT3 磷酸化、STAT3 活性、STAT3 核易位、心肌 IL-10 水平以及 M2c 巨噬细胞的百分比,并减少肌成纤维细胞浸润。LY294002 或 S3I-201 逆转了丁烯基苯酞对 IL-10 水平增加的作用。此外,LY294002 抑制了 STAT3 磷酸化,但不影响 PI3K 活性。总之,宿主环境负责 MI 后与衰老相关的肌成纤维细胞失调,通过靶向 PI3K/STAT3 轴使巨噬细胞向 M2 表型分化,用丁烯基苯酞治疗可以改善这种失调。