Department of Bioengineering, Harbin Institute of Technology, Weihai, 264209 Shandong China.
Hereditas. 2019 Jul 16;156:25. doi: 10.1186/s41065-019-0101-0. eCollection 2019.
Alzheimer's disease (AD) is known to be caused by multiple factors, meanwhile the pathogenic mechanism and development of AD associate closely with genetic factors. Existing understanding of the molecular mechanisms underlying AD remains incomplete.
Gene expression data (GSE48350) derived from post-modern brain was extracted from the Gene Expression Omnibus (GEO) database. The differentially expressed genes (DEGs) were derived from hippocampus and entorhinal cortex regions between AD patients and healthy controls and detected via Morpheus. Functional enrichment analyses, including Gene Ontology (GO) and pathway analyses of DEGs, were performed via Cytoscape and followed by the construction of protein-protein interaction (PPI) network. Hub proteins were screened using the criteria: nodes degree≥10 (for hippocampus tissues) and ≥ 8 (for entorhinal cortex tissues). Molecular Complex Detection (MCODE) was used to filtrate the important clusters. University of California Santa Cruz (UCSC) and the database of RNA-binding protein specificities (RBPDB) were employed to identify the RNA-binding proteins of the long non-coding RNA (lncRNA).
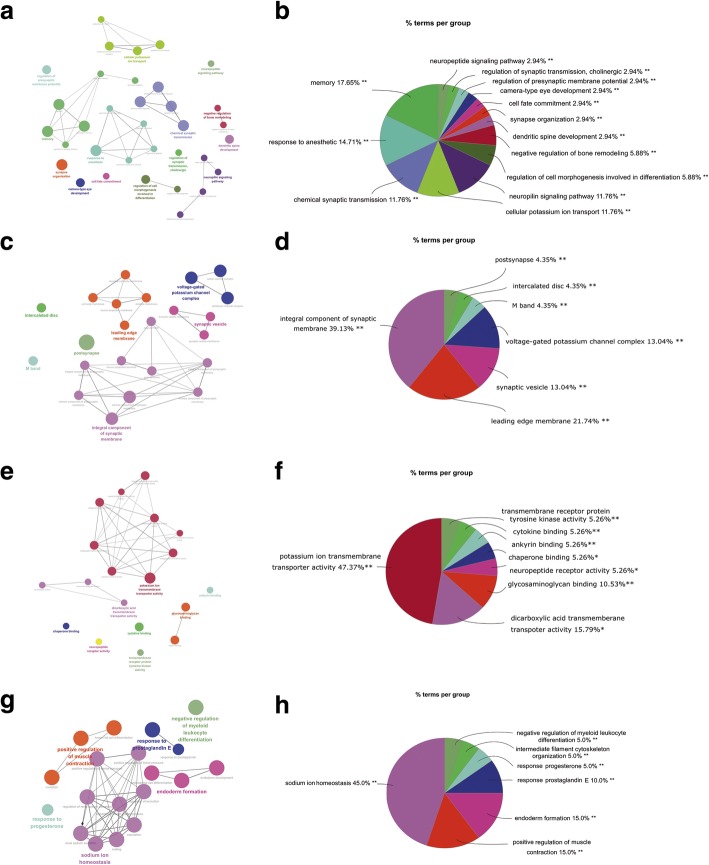
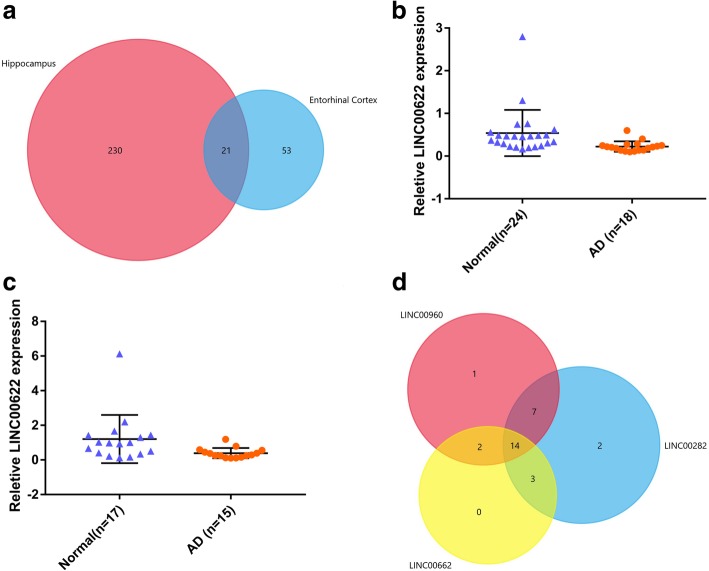
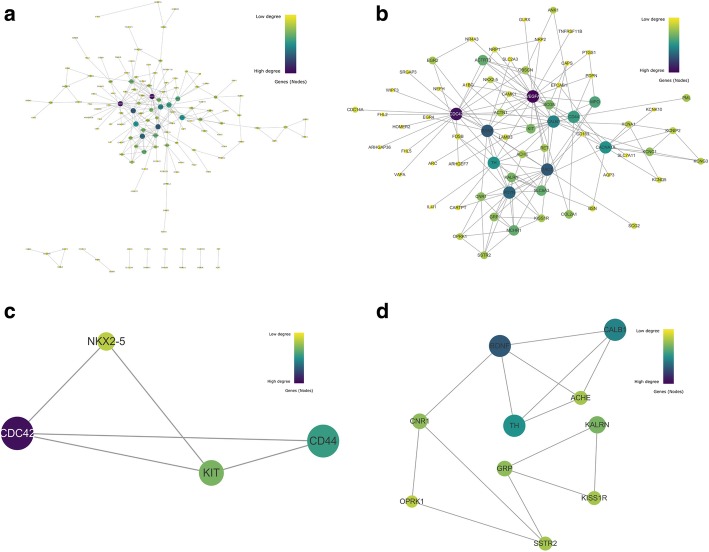
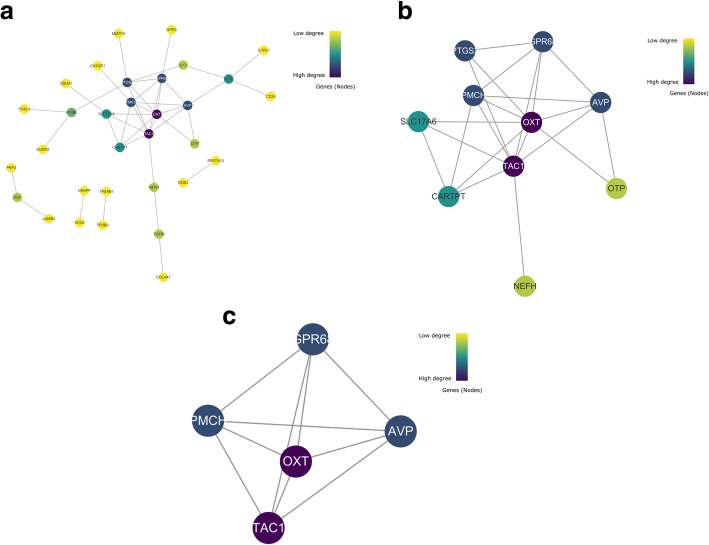
251 & 74 genes were identified as DEGs, which consisted of 56 & 16 up-regulated genes and 195 & 58 down-regulated genes in hippocampus and entorhinal cortex, respectively. Biological analyses demonstrated that the biological processes and pathways related to memory, transmembrane transport, synaptic transmission, neuron survival, drug metabolism, ion homeostasis and signal transduction were enriched in these genes. 11 genes were identified as hub genes in hippocampus and entorhinal cortex, and 3 hub genes were identified as the novel candidates involved in the pathology of AD. Furthermore, 3 lncRNAs were filtrated, whose binding proteins were closely associated with AD.
Through GO enrichment analyses, pathway analyses and PPI analyses, we showed a comprehensive interpretation of the pathogenesis of AD at a systematic biology level, and 3 novel candidate genes and 3 lncRNAs were identified as novel and potential candidates participating in the pathology of AD. The results of this study could supply integrated insights for understanding the pathogenic mechanism underlying AD and potential novel therapeutic targets.
阿尔茨海默病(AD)已知是由多种因素引起的,同时 AD 的发病机制和发展与遗传因素密切相关。目前对 AD 潜在分子机制的认识仍不完整。
从基因表达综合数据库(GEO)中提取来自近代脑组织的基因表达数据(GSE48350)。通过 Morpheus 从 AD 患者和健康对照的海马体和内嗅皮层区域中检测到差异表达基因(DEGs)。通过 Cytoscape 对 DEGs 进行基因本体论(GO)和途径分析等功能富集分析,并构建蛋白质-蛋白质相互作用(PPI)网络。使用节点度≥10(海马体组织)和≥8(内嗅皮层组织)的标准筛选出核心蛋白。使用分子复合物检测(MCODE)过滤重要簇。使用加利福尼亚大学圣克鲁斯分校(UCSC)和 RNA 结合蛋白特异性数据库(RBPDB)鉴定长链非编码 RNA(lncRNA)的 RNA 结合蛋白。
在海马体和内嗅皮层中分别鉴定出 251 个和 74 个 DEGs,其中包括 56 个和 16 个上调基因以及 195 个和 58 个下调基因。生物分析表明,与记忆、跨膜运输、突触传递、神经元存活、药物代谢、离子稳态和信号转导相关的生物学过程和途径在这些基因中得到了富集。在海马体和内嗅皮层中分别鉴定出 11 个和 3 个核心基因作为 AD 病理学的新候选基因。此外,筛选出 3 个 lncRNA,其结合蛋白与 AD 密切相关。
通过 GO 富集分析、途径分析和 PPI 分析,我们在系统生物学水平上对 AD 的发病机制进行了全面的解读,并鉴定出 3 个新的候选基因和 3 个 lncRNA 作为参与 AD 病理学的新的潜在候选基因。本研究结果可为理解 AD 潜在发病机制和潜在新的治疗靶点提供综合见解。