Department of Biology, Saint Louis University, Saint Louis, MO, 63103, USA.
Program in Bioinformatics and Computational Biology, Saint Louis University, Saint Louis, MO, 63103, USA.
Biol Direct. 2019 Aug 1;14(1):12. doi: 10.1186/s13062-019-0242-0.
Metagenomics is the application of modern genomic techniques to investigate the members of a microbial community directly in their natural environments and is widely used in many studies to survey the communities of microbial organisms that live in diverse ecosystems. In order to understand the metagenomic profile of one of the densest interaction spaces for millions of people, the public transit system, the MetaSUB international Consortium has collected and sequenced metagenomes from subways of different cities across the world. In collaboration with CAMDA, MetaSUB has made the metagenomic samples from these cities available for an open challenge of data analysis including, but not limited in scope to, the identification of unknown samples.
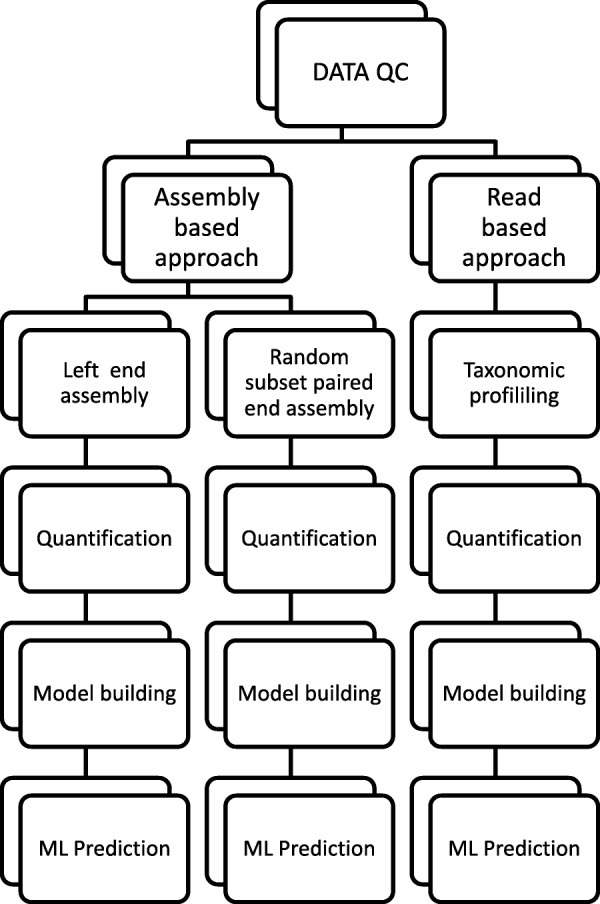
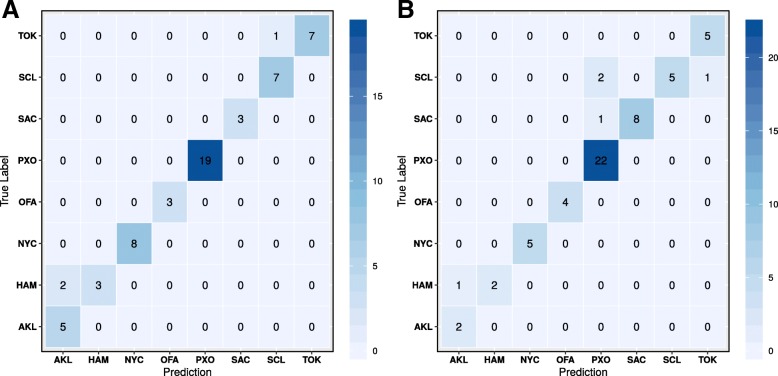
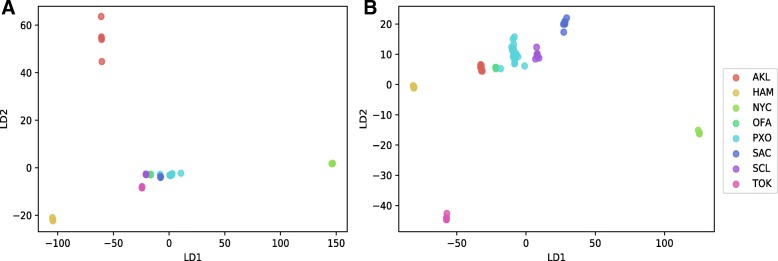
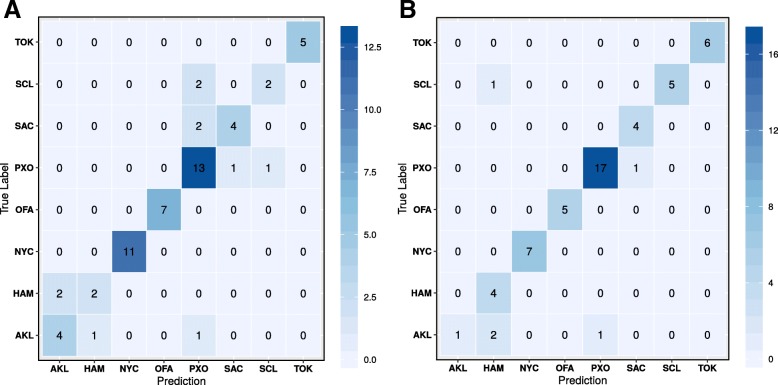
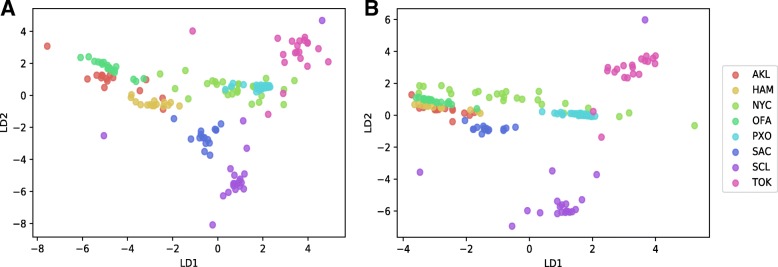
To distinguish the metagenomic profiling among different cities and also predict unknown samples precisely based on the profiling, two different approaches are proposed using machine learning techniques; one is a read-based taxonomy profiling of each sample and prediction method, and the other is a reduced representation assembly-based method. Among various machine learning techniques tested, the random forest technique showed promising results as a suitable classifier for both approaches. Random forest models developed from read-based taxonomic profiling could achieve an accuracy of 91% with 95% confidence interval between 80 and 93%. The assembly-based random forest model prediction also reached 90% accuracy. However, both models achieved roughly the same accuracy on the testing test, whereby they both failed to predict the most abundant label.
Our results suggest that both read-based and assembly-based approaches are powerful tools for the analysis of metagenomics data. Moreover, our results suggest that reduced representation assembly-based methods are able to simultaneous provide high-accuracy prediction on available data. Overall, we show that metagenomic samples can be traced back to their location with careful generation of features from the composition of microbes and utilizing existing machine learning algorithms. Proposed approaches show high accuracy of prediction, but require careful inspection before making any decisions due to sample noise or complexity.
This article was reviewed by Eugene V. Koonin, Jing Zhou and Serghei Mangul.
宏基因组学是将现代基因组技术应用于直接在自然环境中研究微生物群落成员的学科,广泛用于许多研究中,以调查生活在各种生态系统中的微生物生物群落。为了了解数以百万计的人密集互动空间之一——公共交通系统的宏基因组特征,MetaSUB 国际联盟已经从世界各地不同城市的地铁中收集和测序了宏基因组。与 CAMDA 合作,MetaSUB 为数据分析的公开挑战提供了来自这些城市的宏基因组样本,包括但不限于识别未知样本。
为了区分不同城市的宏基因组特征,并根据特征准确预测未知样本,我们使用机器学习技术提出了两种不同的方法;一种是基于每个样本的基于读取的分类学特征分析和预测方法,另一种是基于简化表示的组装方法。在测试的各种机器学习技术中,随机森林技术表现出作为两种方法合适分类器的有前途的结果。基于基于读取的分类学特征分析的随机森林模型可以达到 91%的准确率,置信区间为 80%至 93%。基于组装的随机森林模型预测也达到了 90%的准确率。然而,两种模型在测试中都达到了大致相同的准确率,都无法预测最丰富的标签。
我们的结果表明,基于读取和基于组装的方法都是分析宏基因组数据的有力工具。此外,我们的结果表明,基于简化表示的组装方法能够同时对现有数据进行高精度预测。总体而言,我们表明,通过仔细生成微生物组成的特征并利用现有的机器学习算法,可以追溯到宏基因组样本的来源。所提出的方法具有很高的预测准确性,但由于样本噪声或复杂性,在做出任何决策之前需要仔细检查。
本文由 Eugene V. Koonin、Jing Zhou 和 Serghei Mangul 进行了评审。