Lewis Scott, Nash Andrea, Li Qinghong, Ahn Tae-Hyuk
Program in Bioinformatics and Computational Biology, Saint Louis University, St. Louis, MO, USA.
Nestlé Purina Research, St. Louis, MO, USA.
BioData Min. 2021 Aug 21;14(1):41. doi: 10.1186/s13040-021-00270-x.
Recent advances in sequencing technologies have driven studies identifying the microbiome as a key regulator of overall health and disease in the host. Both 16S amplicon and whole genome shotgun sequencing technologies are currently being used to investigate this relationship, however, the choice of sequencing technology often depends on the nature and experimental design of the study. In principle, the outputs rendered by analysis pipelines are heavily influenced by the data used as input; it is then important to consider that the genomic features produced by different sequencing technologies may emphasize different results.
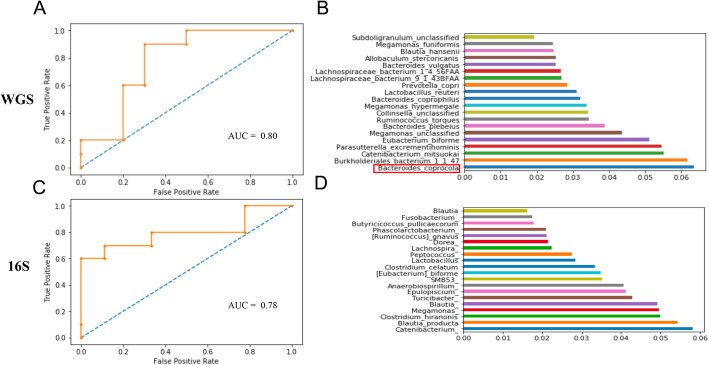
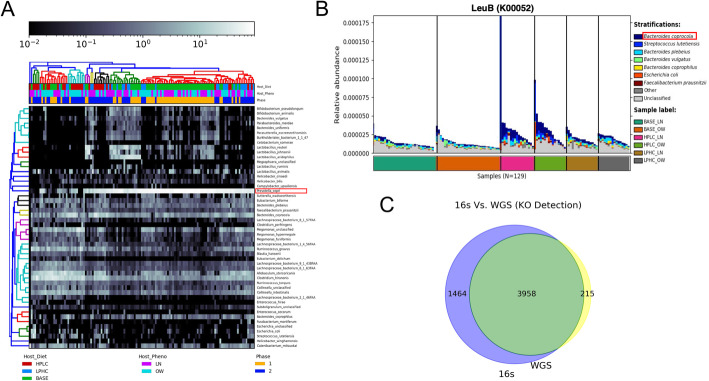
In this work, we use public 16S amplicon and whole genome shotgun sequencing (WGS) data from the same dogs to investigate the relationship between sequencing technology and the captured gut metagenomic landscape in dogs. In our analyses, we compare the taxonomic resolution at the species and phyla levels and benchmark 12 classification algorithms in their ability to accurately identify host phenotype using only taxonomic relative abundance information from 16S and WGS datasets with identical study designs. Our best performing model, a random forest trained by the WGS dataset, identified a species (Bacteroides coprocola) that predominantly contributes to the abundance of leuB, a gene involved in branched chain amino acid biosynthesis; a risk factor for glucose intolerance, insulin resistance, and type 2 diabetes. This trend was not conserved when we trained the model using 16S sequencing profiles from the same dogs.
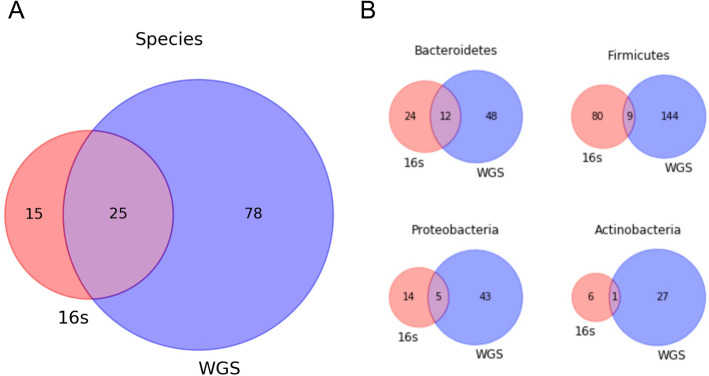
Our results indicate that WGS sequencing of dog microbiomes detects a greater taxonomic diversity than 16S sequencing of the same dogs at the species level and with respect to four gut-enriched phyla levels. This difference in detection does not significantly impact the performance metrics of machine learning algorithms after down-sampling. Although the important features extracted from our best performing model are not conserved between the two technologies, the important features extracted from either instance indicate the utility of machine learning algorithms in identifying biologically meaningful relationships between the host and microbiome community members. In conclusion, this work provides the first systematic machine learning comparison of dog 16S and WGS microbiomes derived from identical study designs.
测序技术的最新进展推动了相关研究,这些研究将微生物组确定为宿主整体健康和疾病的关键调节因子。目前,16S扩增子测序技术和全基因组鸟枪法测序技术都被用于研究这种关系,然而,测序技术的选择通常取决于研究的性质和实验设计。原则上,分析流程产生的输出结果在很大程度上受到用作输入的数据的影响;因此,重要的是要考虑到不同测序技术产生的基因组特征可能会强调不同的结果。
在这项工作中,我们使用来自相同犬只的公开16S扩增子测序数据和全基因组鸟枪法测序(WGS)数据,来研究测序技术与犬只肠道宏基因组景观之间的关系。在我们的分析中,我们比较了物种和门水平的分类分辨率,并对12种分类算法进行基准测试,这些算法仅使用来自具有相同研究设计的16S和WGS数据集的分类相对丰度信息来准确识别宿主表型的能力。我们表现最佳的模型是一个由WGS数据集训练的随机森林模型,它识别出一种物种(粪便拟杆菌),该物种主要导致亮氨酸B(一种参与支链氨基酸生物合成的基因)丰度升高;亮氨酸B是葡萄糖不耐受、胰岛素抵抗和2型糖尿病的一个风险因素。当我们使用来自相同犬只的16S测序图谱训练模型时,这种趋势并不一致。
我们的结果表明,犬只微生物组的WGS测序在物种水平以及四个肠道富集门水平上,比相同犬只的16S测序检测到更大的分类多样性。在进行下采样后,这种检测差异对机器学习算法的性能指标没有显著影响。尽管从我们表现最佳的模型中提取的重要特征在两种技术之间并不一致,但从任何一种情况中提取的重要特征都表明了机器学习算法在识别宿主与微生物群落成员之间生物学上有意义的关系方面的效用。总之,这项工作提供了首次对源自相同研究设计的犬只16S和WGS微生物组进行的系统机器学习比较。