From the Department of Medicine, Division of Cardiology (M.S.S., R.A.B., M.B.F., A.S.R., B.Y.E., K.A.K., M.A.C., K.S., M.P.Y.L., T.A.M.), University of Colorado Anschutz Medical Campus, Aurora.
Consortium for Fibrosis Research & Translation (M.S.S., R.A.B., M.B.F., A.S.R., B.Y.E., K.A.K., M.A.C., K.S., M.P.Y.L., T.A.M.), University of Colorado Anschutz Medical Campus, Aurora.
Circ Res. 2019 Sep 13;125(7):662-677. doi: 10.1161/CIRCRESAHA.119.315125. Epub 2019 Aug 14.
Small molecule inhibitors of the acetyl-histone binding protein BRD4 have been shown to block cardiac fibrosis in preclinical models of heart failure (HF). However, since the inhibitors target BRD4 ubiquitously, it is unclear whether this chromatin reader protein functions in cell type-specific manner to control pathological myocardial fibrosis. Furthermore, the molecular mechanisms by which BRD4 stimulates the transcriptional program for cardiac fibrosis remain unknown.
We sought to test the hypothesis that BRD4 functions in a cell-autonomous and signal-responsive manner to control activation of cardiac fibroblasts, which are the major extracellular matrix-producing cells of the heart.
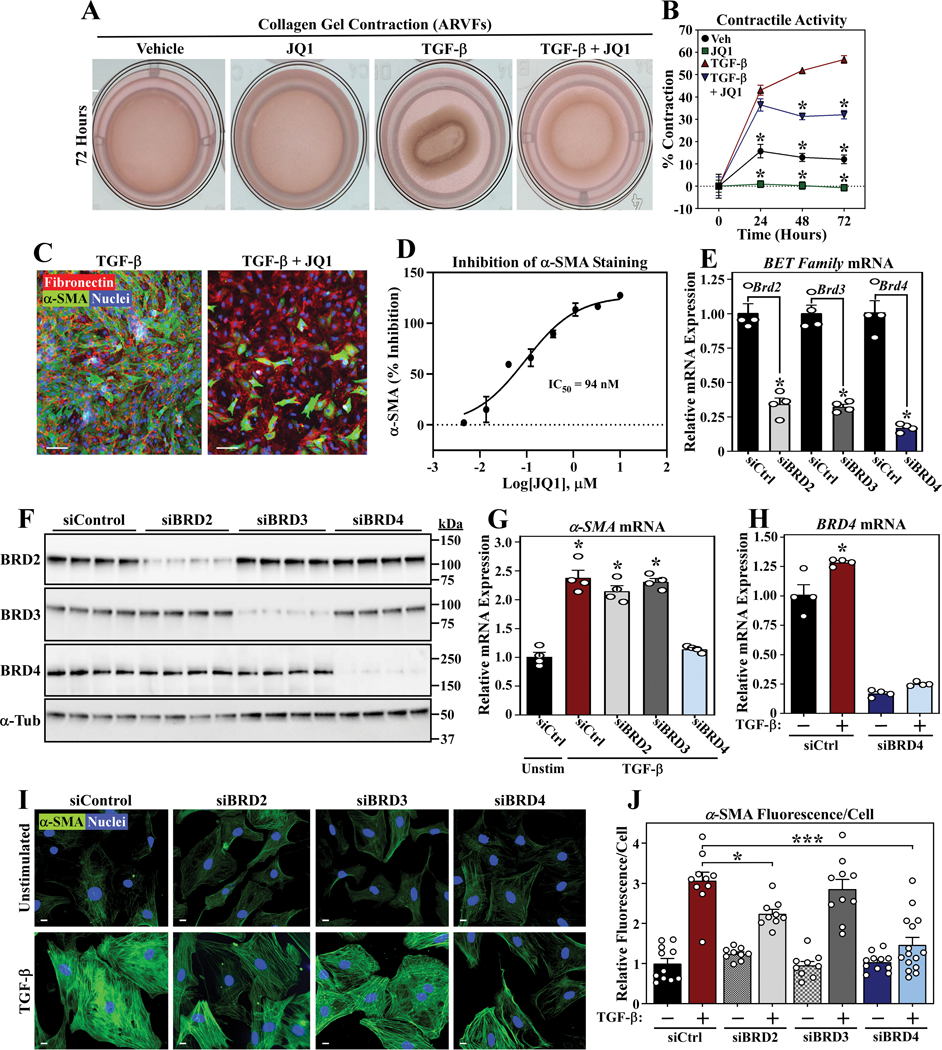
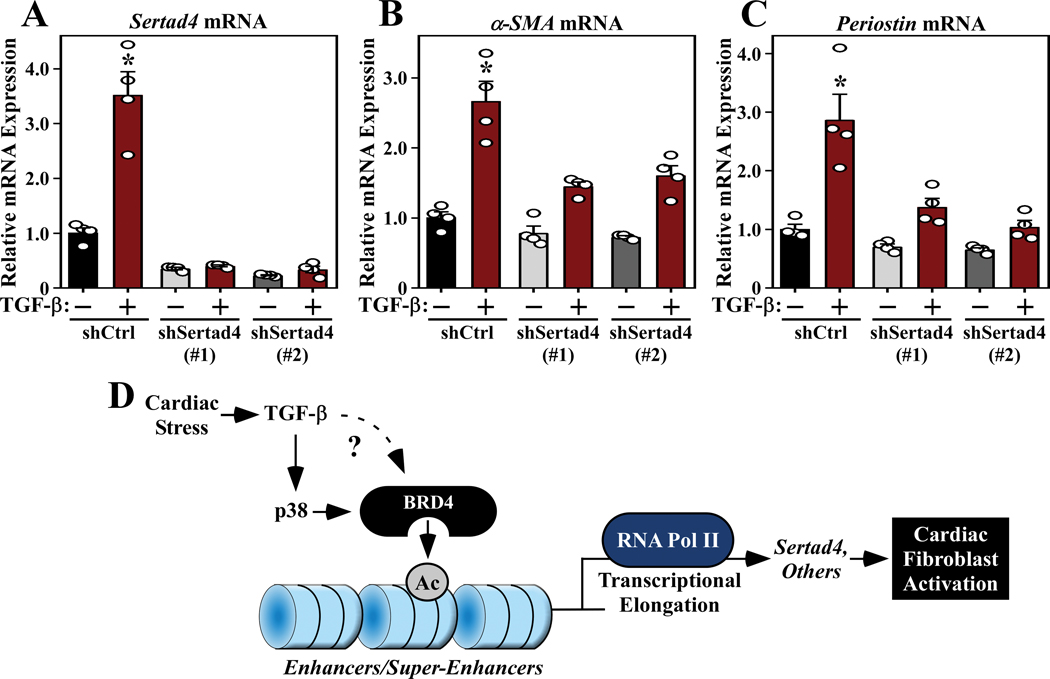
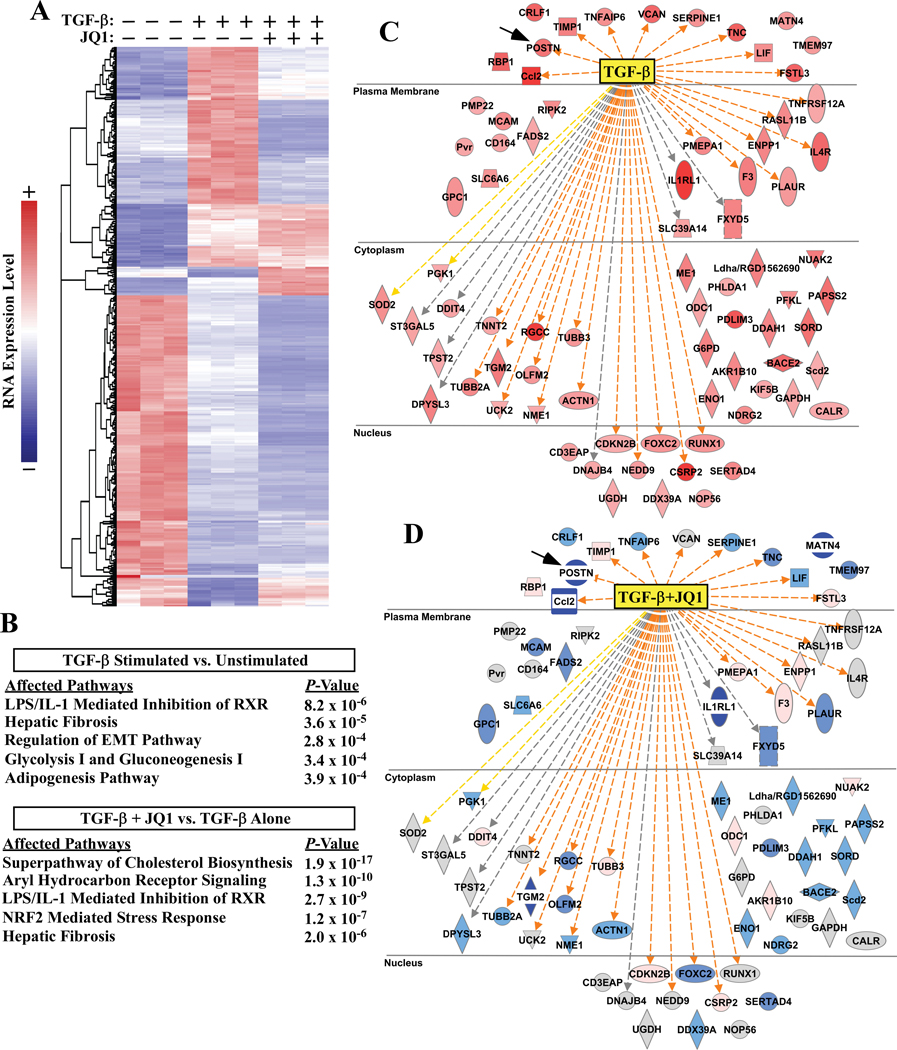
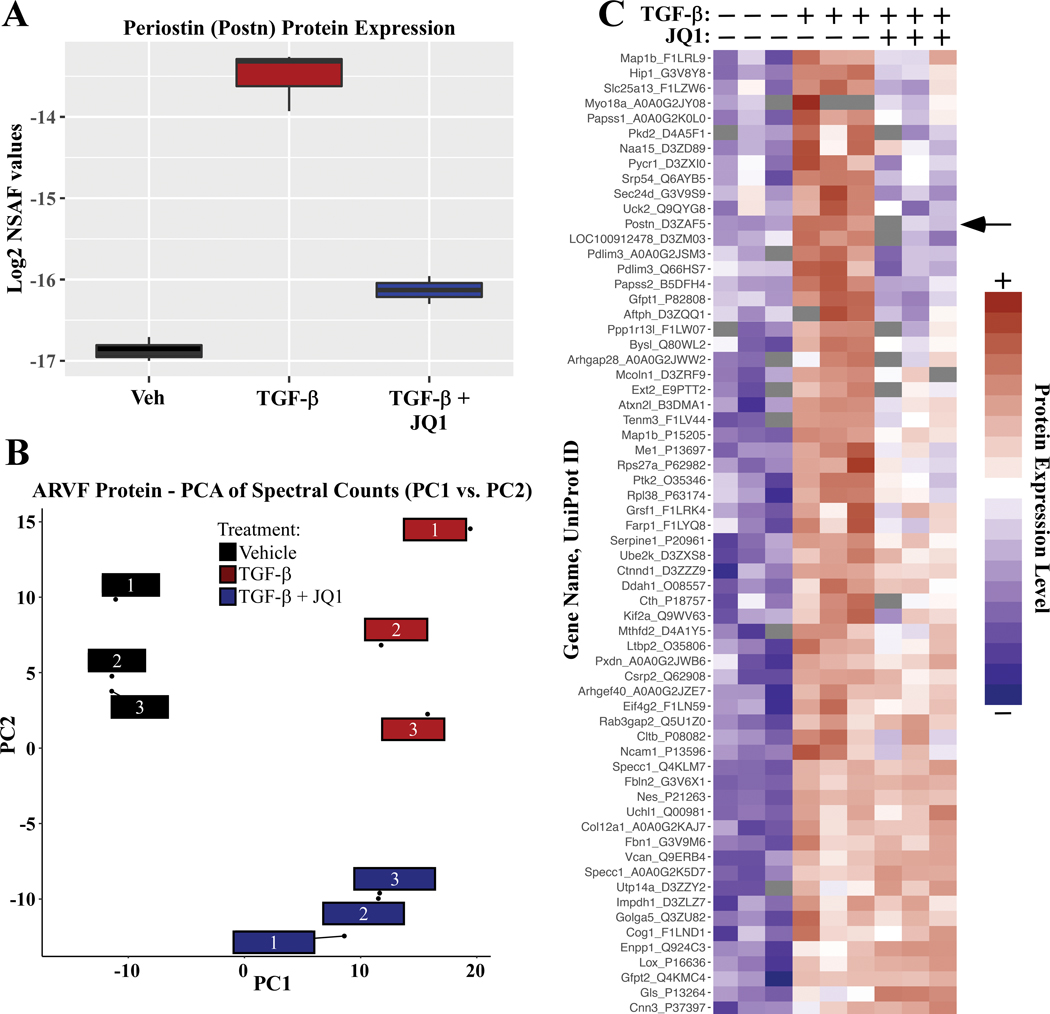
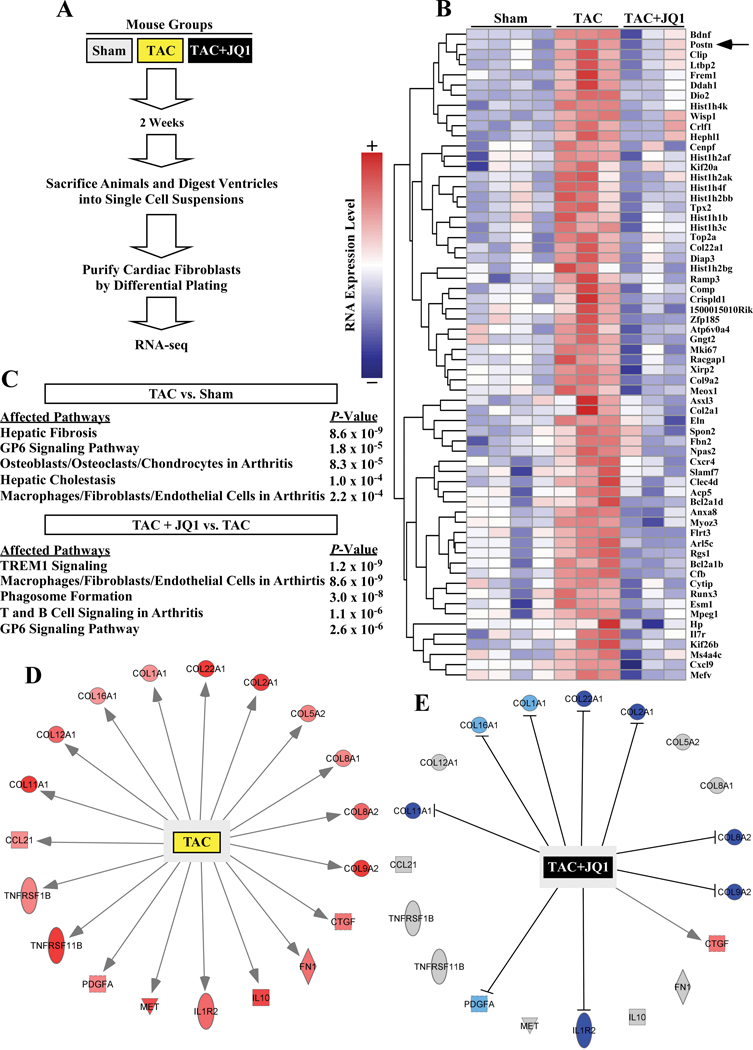
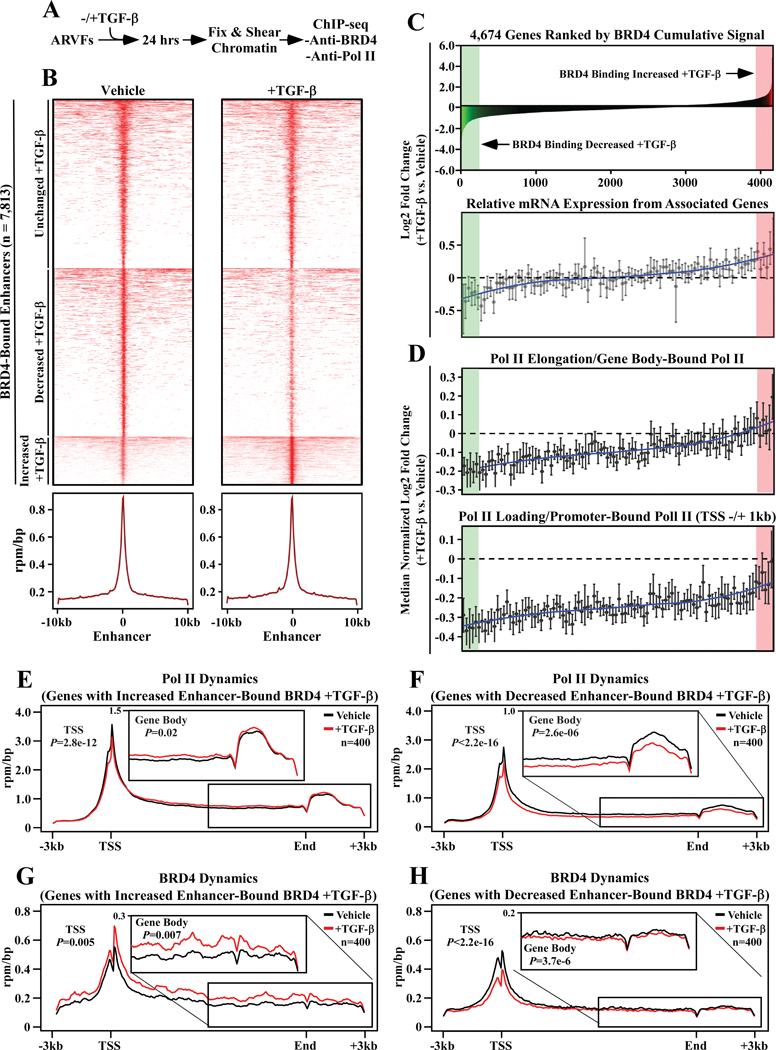
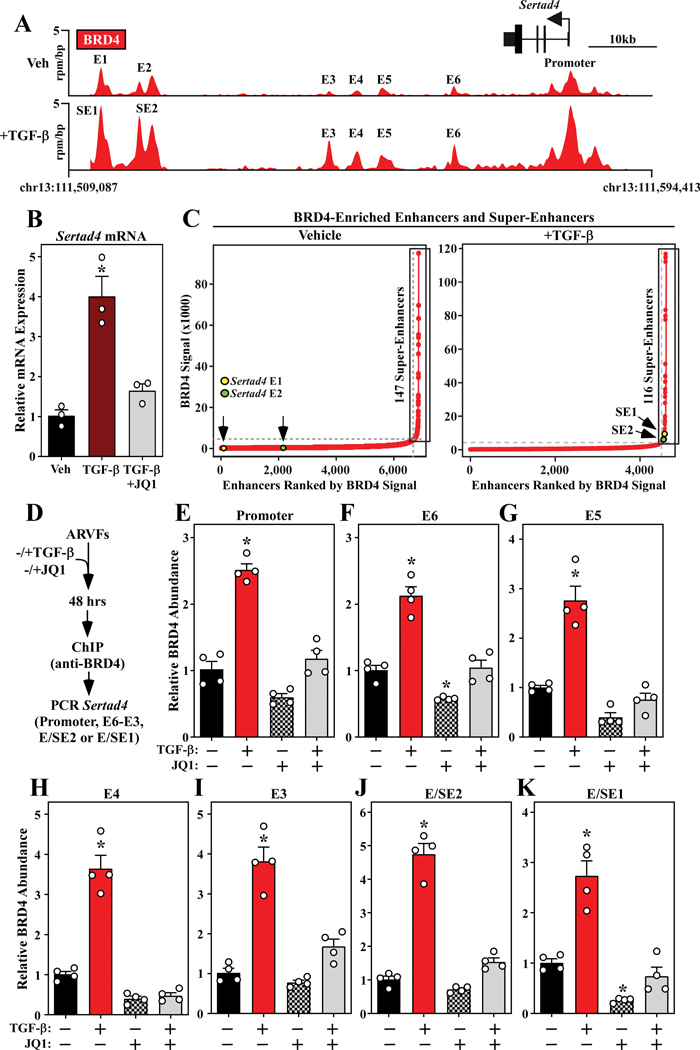
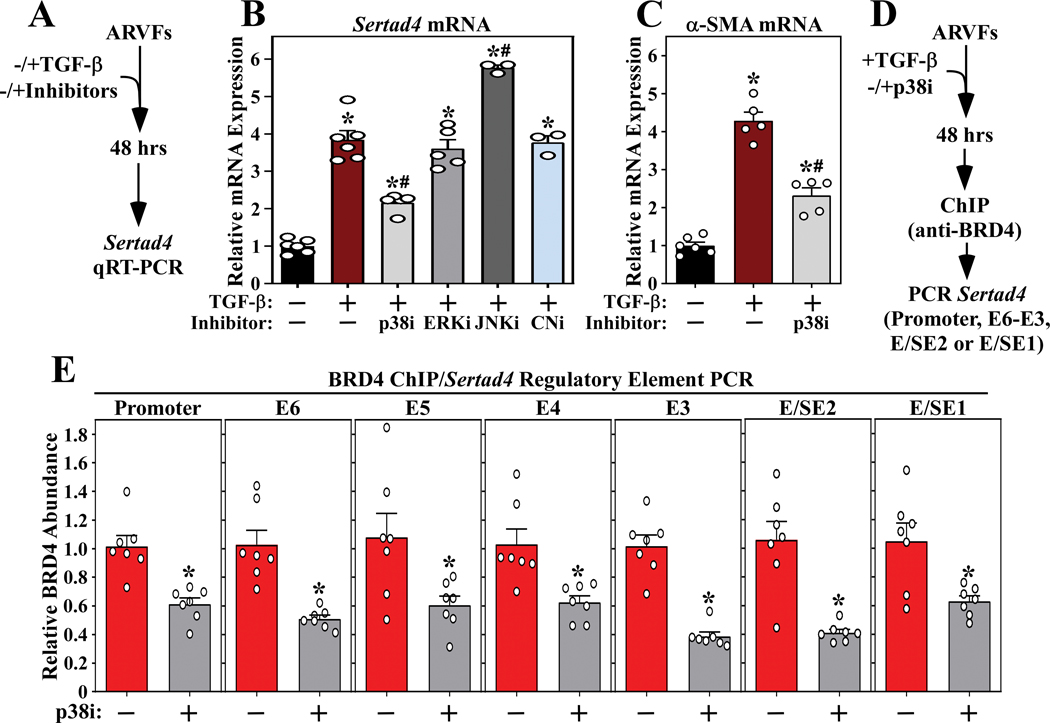
RNA-sequencing, mass spectrometry, and cell-based assays employing primary adult rat ventricular fibroblasts demonstrated that BRD4 functions as an effector of TGF-β (transforming growth factor-β) signaling to stimulate conversion of quiescent cardiac fibroblasts into ()-positive cells that express high levels of extracellular matrix. These findings were confirmed in vivo through whole-transcriptome analysis of cardiac fibroblasts from mice subjected to transverse aortic constriction and treated with the small molecule BRD4 inhibitor, JQ1. Chromatin immunoprecipitation-sequencing revealed that BRD4 undergoes stimulus-dependent, genome-wide redistribution in cardiac fibroblasts, becoming enriched on a subset of enhancers and super-enhancers, and leading to RNA polymerase II activation and expression of downstream target genes. Employing the (SERTA domain-containing protein 4) locus as a prototype, we demonstrate that dynamic chromatin targeting of BRD4 is controlled, in part, by p38 MAPK (mitogen-activated protein kinase) and provide evidence of a critical function for in TGF-β-mediated cardiac fibroblast activation.
These findings define BRD4 as a central regulator of the pro-fibrotic cardiac fibroblast phenotype, establish a p38-dependent signaling circuit for epigenetic reprogramming in heart failure, and uncover a novel role for . The work provides a mechanistic foundation for the development of BRD4 inhibitors as targeted anti-fibrotic therapies for the heart.
小分子乙酰组蛋白结合蛋白 BRD4 抑制剂已被证明可在心力衰竭 (HF) 的临床前模型中阻断心脏纤维化。然而,由于抑制剂普遍靶向 BRD4,尚不清楚这种染色质读蛋白是否以细胞类型特异性方式发挥作用来控制病理性心肌纤维化。此外,BRD4 刺激心脏纤维化转录程序的分子机制尚不清楚。
我们试图检验以下假设,即 BRD4 以自主和信号响应的方式发挥作用,以控制心肌成纤维细胞的激活,心肌成纤维细胞是心脏的主要细胞外基质产生细胞。
使用原代成年大鼠心室成纤维细胞进行 RNA 测序、质谱分析和基于细胞的测定表明,BRD4 作为 TGF-β(转化生长因子-β)信号的效应物发挥作用,刺激静止的心肌成纤维细胞转化为 ()-阳性细胞,这些细胞表达高水平的细胞外基质。这些发现通过对接受横主动脉缩窄和接受小分子 BRD4 抑制剂 JQ1 治疗的小鼠的心脏成纤维细胞进行全转录组分析得到了证实。染色质免疫沉淀测序显示,BRD4 在心肌成纤维细胞中经历刺激依赖性的全基因组重新分布,在一组增强子和超级增强子上富集,并导致 RNA 聚合酶 II 激活和下游靶基因的表达。我们以 (SERTA 结构域包含蛋白 4) 基因座作为原型,证明 BRD4 的动态染色质靶向部分受 p38 MAPK(丝裂原活化蛋白激酶)控制,并提供了 BRD4 在 TGF-β 介导的心肌成纤维细胞激活中发挥关键功能的证据。
这些发现将 BRD4 定义为促纤维化心肌成纤维细胞表型的核心调节剂,确立了 p38 依赖性信号通路在心力衰竭中的表观遗传重编程,并揭示了 的新作用。这项工作为 BRD4 抑制剂作为心脏靶向抗纤维化疗法的开发提供了机制基础。