Aina Alexander A, Misquitta Alston J, Phipps Maximillian J S, Price Sarah L
Department of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, U.K.
School of Physics and Astronomy and the Thomas Young Centre for Theory and Simulation of Materials at Queen Mary, University of London, London E1 4NS, U.K.
ACS Omega. 2019 May 16;4(5):8614-8625. doi: 10.1021/acsomega.9b00648. eCollection 2019 May 31.
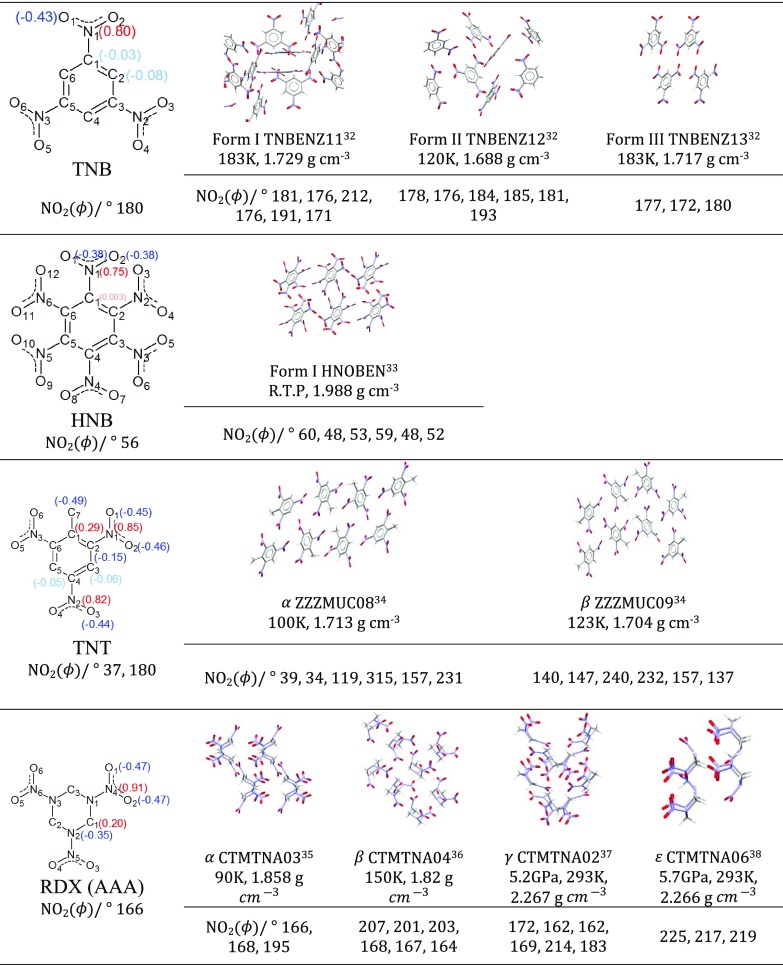
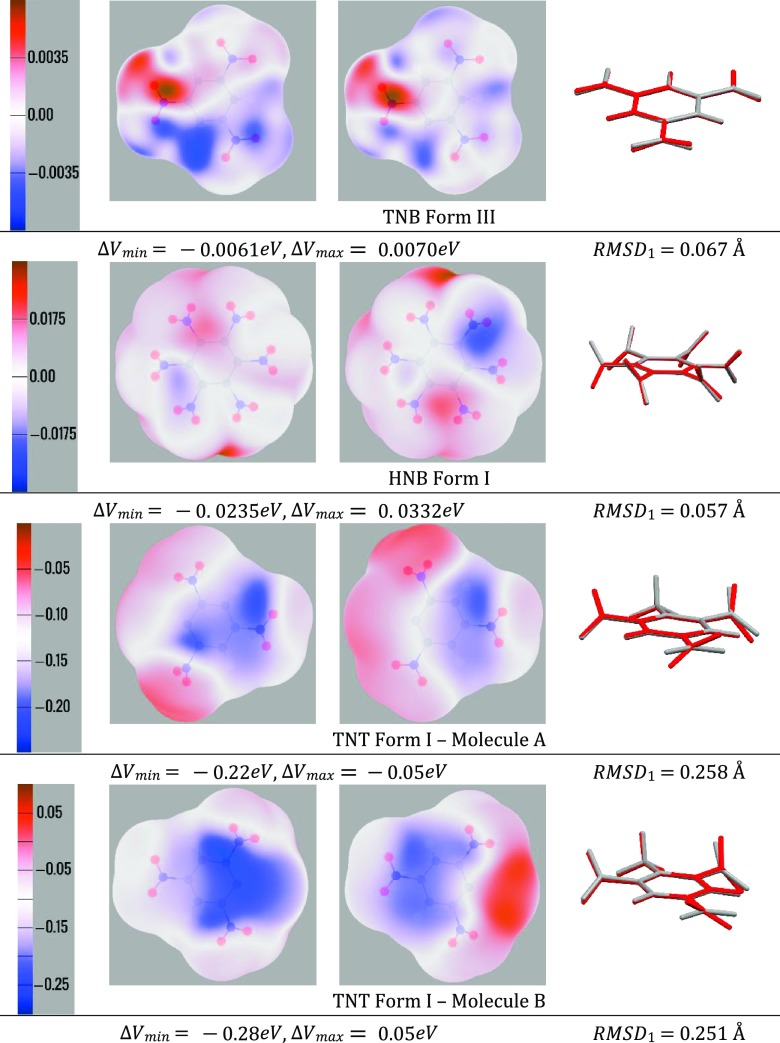
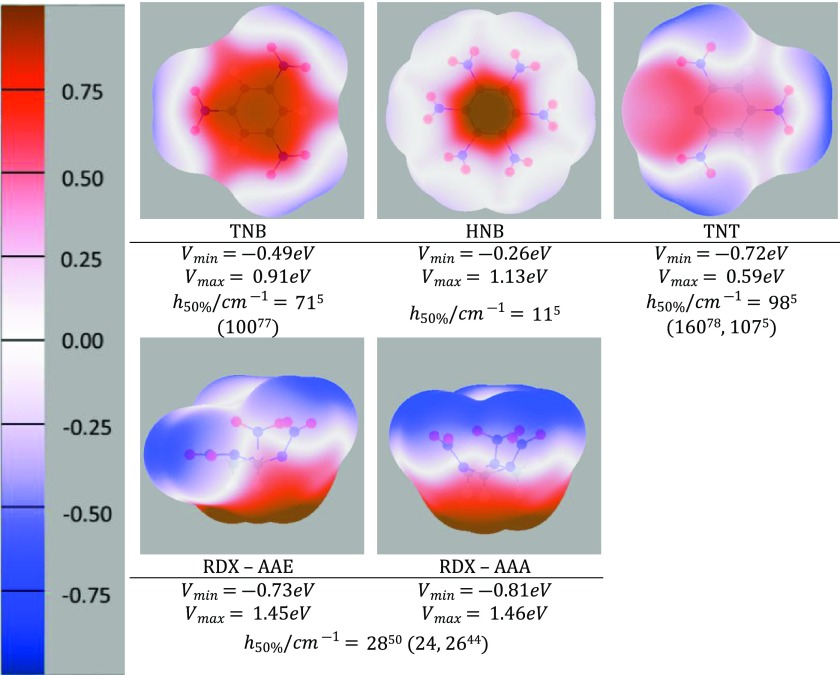
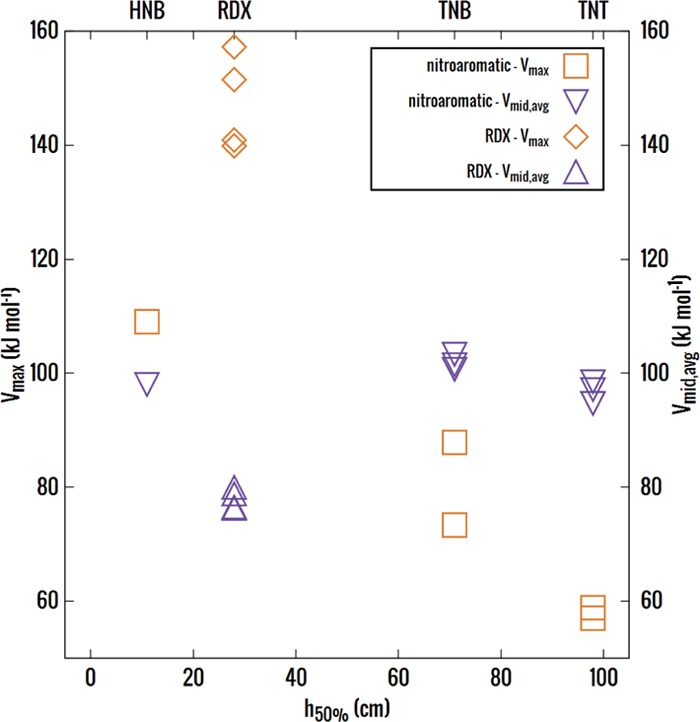
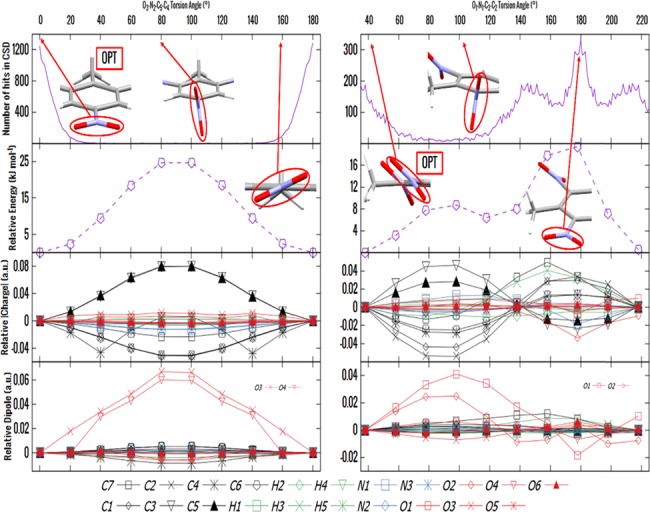
The charge distribution of NO groups within the crystalline polymorphs of energetic materials strongly affects their explosive properties. We use the recently introduced basis-space iterated stockholder atom partitioning of high-quality charge distributions to examine the approximations that can be made in modeling polymorphs and their physical properties, using 1,3,5-trinitroperhydro-1,3,5-triazine, trinitrotoluene, 1-3-5-trinitrobenzene, and hexanitrobenzene as exemplars. The NO charge distribution is strongly affected by the neighboring atoms, the rest of the molecules, and also significantly by the NO torsion angle within the possible variations found in observed crystal structures. Thus, the proposed correlations between the molecular electrostatic properties, such as trigger-bond potential or maxima in the electrostatic potential, and impact sensitivity will be affected by the changes in conformation that occur on crystallization. We establish the relationship between the NO torsion angle and the likelihood of occurrence in observed crystal structures, the conformational energy, and the charge and dipole magnitude on each atom, and how this varies with the neighboring groups. We examine the effect of analytically rotating the atomic multipole moments to model changes in torsion angle and establish that this is a viable approach for crystal structures but is not accurate enough to model the relative lattice energies. This establishes the basis of transferability of the NO charge distribution for realistic nonempirical model intermolecular potentials for simulating energetic materials.
含能材料晶体多晶型物中硝基(NO)基团的电荷分布强烈影响其爆炸性能。我们使用最近引入的高质量电荷分布的基空间迭代股东原子划分方法,以1,3,5-三硝基全氢-1,3,5-三嗪、三硝基甲苯、1,3,5-三硝基苯和六硝基苯为示例,检验在多晶型物及其物理性质建模中可以采用的近似方法。硝基电荷分布受到相邻原子、分子其余部分的强烈影响,并且在观察到的晶体结构中可能的变化范围内,还受到硝基扭转角的显著影响。因此,所提出的分子静电性质(如触发键势或静电势最大值)与撞击感度之间的相关性将受到结晶时发生的构象变化的影响。我们建立了硝基扭转角与在观察到的晶体结构中的出现概率、构象能以及每个原子上的电荷和偶极矩之间的关系,以及这如何随相邻基团而变化。我们研究了通过解析旋转原子多极矩来模拟扭转角变化的效果,并确定这对于晶体结构是一种可行的方法,但对于模拟相对晶格能来说不够精确。这为模拟含能材料的实际非经验模型分子间势建立了硝基电荷分布可转移性的基础。