Geng Hao, Chen Fangfang, Ye Jing, Jiang Fan
Lab of Computational Chemistry and Drug Design, State Key Laboratory of Chemical Oncogenomics, Peking University Shenzhen Graduate School, Shenzhen 518055, China.
Guangdong and Shenzhen Key Laboratory of Male Reproductive Medicine and Genetics, Peking University Shenzhen Hospital, Shenzhen PKU-HKUST Medical Center, Shenzhen 518036, China.
Comput Struct Biotechnol J. 2019 Jul 26;17:1162-1170. doi: 10.1016/j.csbj.2019.07.010. eCollection 2019.
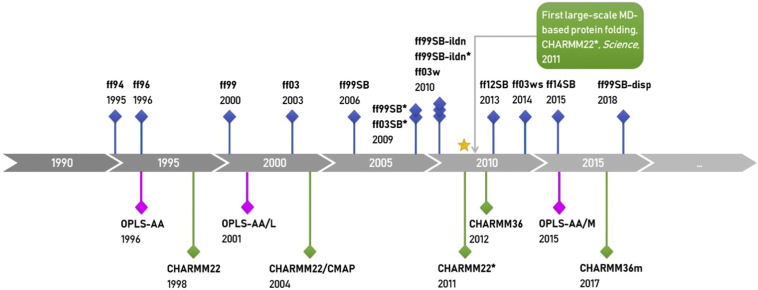
Compared with rapid accumulation of protein sequences from high-throughput DNA sequencing, obtaining experimental 3D structures of proteins is still much more difficult, making protein structure prediction (PSP) potentially very useful. Currently, a vast majority of PSP efforts are based on data mining of known sequences, structures and their relationships (informatics-based). However, if closely related template is not available, these methods are usually much less reliable than experiments. They may also be problematic in predicting the structures of naturally occurring or designed peptides. On the other hand, physics-based methods including molecular dynamics (MD) can utilize our understanding of detailed atomic interactions determining biomolecular structures. In this mini-review, we show that all-atom MD can predict structures of cyclic peptides and other peptide foldamers with accuracy similar to experiments. Then, some notable successes in reproducing experimental 3D structures of small proteins through MD simulations (some with replica-exchange) of the folding were summarized. We also describe advancements of MD-based refinement of structure models, and the integration of limited experimental or bioinformatics data into MD-based structure modeling.
与通过高通量DNA测序快速积累蛋白质序列相比,获取蛋白质的实验性三维结构仍然困难得多,这使得蛋白质结构预测(PSP)可能非常有用。目前,绝大多数的PSP工作是基于对已知序列、结构及其关系的数据挖掘(基于信息学)。然而,如果没有密切相关的模板,这些方法通常比实验的可靠性要低得多。它们在预测天然存在或设计的肽的结构时也可能存在问题。另一方面,包括分子动力学(MD)在内的基于物理学的方法可以利用我们对决定生物分子结构的详细原子相互作用的理解。在本综述中,我们表明全原子MD可以以与实验相似的精度预测环肽和其他肽折叠体的结构。然后,总结了通过折叠的MD模拟(一些采用副本交换)在重现小蛋白质的实验性三维结构方面取得的一些显著成功。我们还描述了基于MD的结构模型优化的进展,以及将有限的实验或生物信息学数据整合到基于MD的结构建模中。