Department of Pediatrics, Women and Infants Hospital of RI, Providence, RI, USA.
Department of Pediatrics, Brown University Warren Alpert Medical School, Providence, RI, USA.
Sci Rep. 2019 Sep 2;9(1):12648. doi: 10.1038/s41598-019-49114-z.
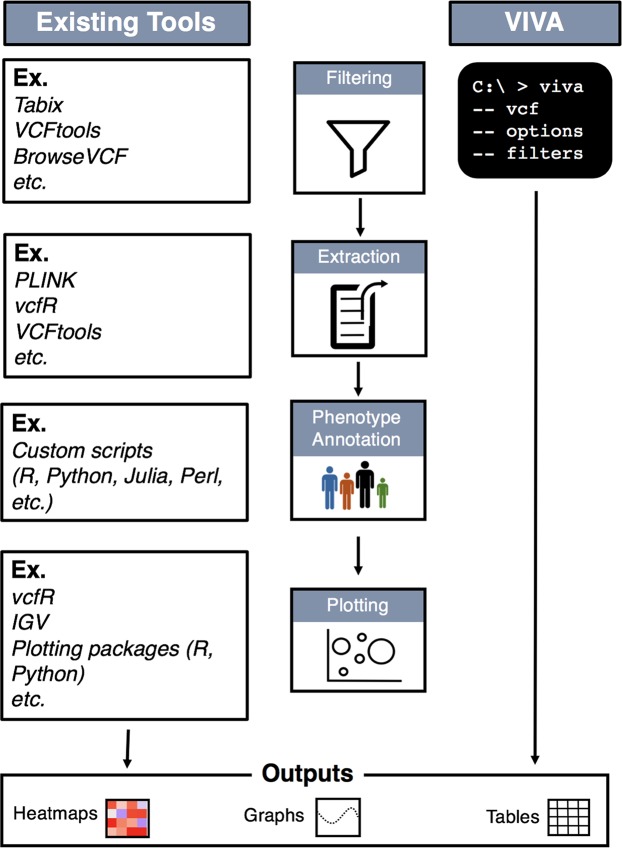
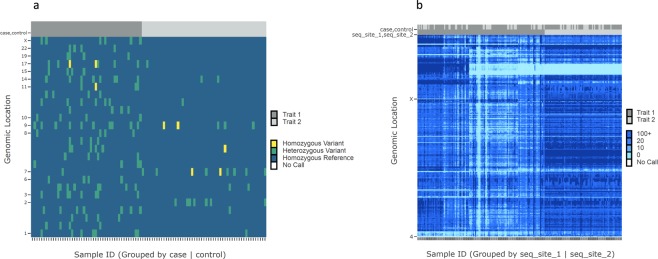
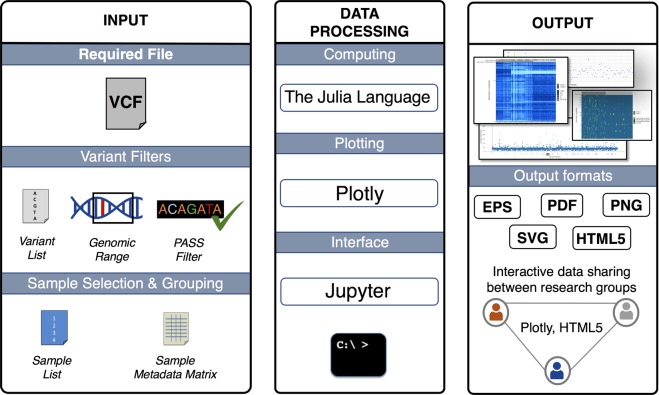
High-throughput sequencing produces an extraordinary amount of genomic data that is organized into a number of high-dimension datasets. Accordingly, visualization of genomic data has become essential for quality control, exploration, and data interpretation. The Variant Call Format (VCF) is a text file format generated during the variant calling process that contains genomic information and locations of variants in a group of sequenced samples. The current workflow for visualization of genomic variant data from VCF files requires use of a combination of existing tools. Here, we describe VIVA (VIsualization of VAriants), a command line utility and Jupyter Notebook based tool for evaluating and sharing genomic data for variant analysis and quality control of sequencing experiments from VCF files. VIVA combines the functionality of existing tools into a single command to interactively evaluate and share genomic data, as well as create publication quality graphics.
高通量测序产生了大量的基因组数据,这些数据被组织成了许多高维数据集。因此,基因组数据的可视化对于质量控制、探索和数据解释变得至关重要。变体调用格式 (VCF) 是在变体调用过程中生成的一种文本文件格式,其中包含了一组测序样本中基因组信息和变体位置。目前,从 VCF 文件可视化基因组变体数据的工作流程需要结合使用现有的工具。在这里,我们描述了 VIVA(变体可视化),这是一个基于命令行实用程序和 Jupyter Notebook 的工具,用于评估和共享基因组数据,用于从 VCF 文件进行变体分析和测序实验的质量控制。VIVA 将现有工具的功能组合到一个命令中,以便交互式地评估和共享基因组数据,并创建具有出版质量的图形。