Diaz-Mejia J Javier, Meng Elaine C, Pico Alexander R, MacParland Sonya A, Ketela Troy, Pugh Trevor J, Bader Gary D, Morris John H
Princess Margaret Cancer Centre, University Health Network, Toronto, ON, M5G 2M9, Canada.
The Donnelly Centre, University of Toronto, Toronto, ON, M5S 3E1, Canada.
F1000Res. 2019 Mar 15;8. doi: 10.12688/f1000research.18490.3. eCollection 2019.
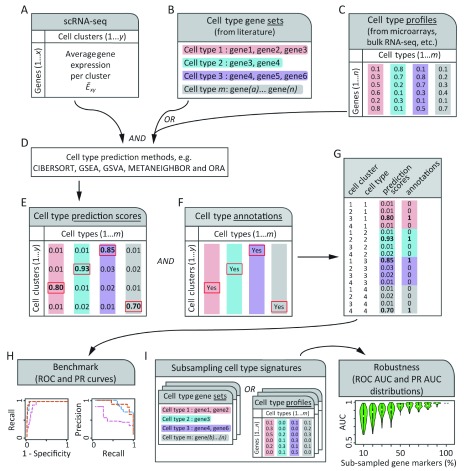
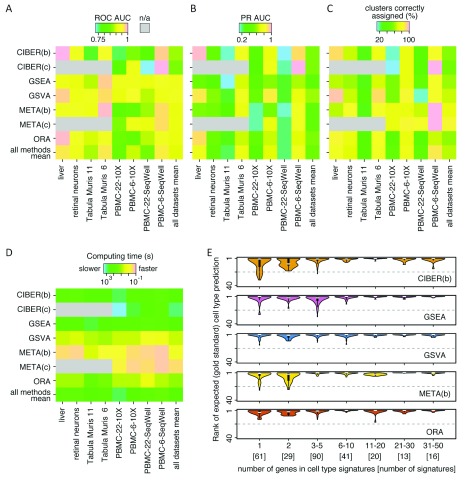
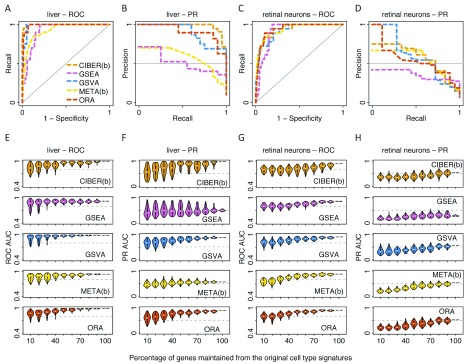
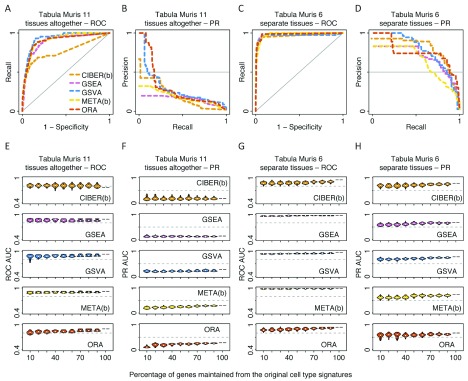
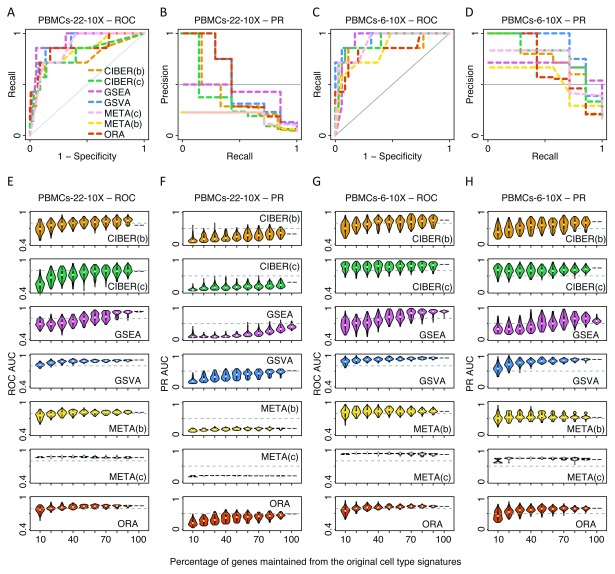
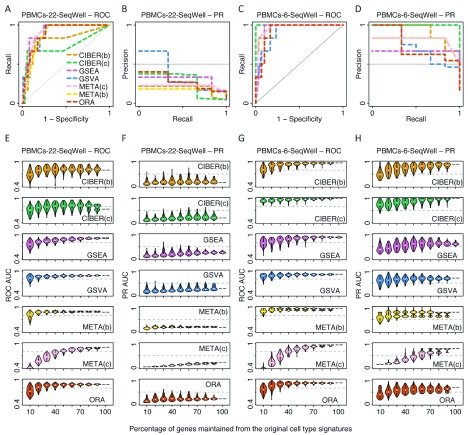
Identification of cell type subpopulations from complex cell mixtures using single-cell RNA-sequencing (scRNA-seq) data includes automated steps from normalization to cell clustering. However, assigning cell type labels to cell clusters is often conducted manually, resulting in limited documentation, low reproducibility and uncontrolled vocabularies. This is partially due to the scarcity of reference cell type signatures and because some methods support limited cell type signatures. In this study, we benchmarked five methods representing first-generation enrichment analysis (ORA), second-generation approaches (GSEA and GSVA), machine learning tools (CIBERSORT) and network-based neighbor voting (METANEIGHBOR), for the task of assigning cell type labels to cell clusters from scRNA-seq data. We used five scRNA-seq datasets: human liver, 11 Tabula Muris mouse tissues, two human peripheral blood mononuclear cell datasets, and mouse retinal neurons, for which reference cell type signatures were available. The datasets span Drop-seq, 10X Chromium and Seq-Well technologies and range in size from ~3,700 to ~68,000 cells. Our results show that, in general, all five methods perform well in the task as evaluated by receiver operating characteristic curve analysis (average area under the curve (AUC) = 0.91, sd = 0.06), whereas precision-recall analyses show a wide variation depending on the method and dataset (average AUC = 0.53, sd = 0.24). We observed an influence of the number of genes in cell type signatures on performance, with smaller signatures leading more frequently to incorrect results. GSVA was the overall top performer and was more robust in cell type signature subsampling simulations, although different methods performed well using different datasets. METANEIGHBOR and GSVA were the fastest methods. CIBERSORT and METANEIGHBOR were more influenced than the other methods by analyses including only expected cell types. We provide an extensible framework that can be used to evaluate other methods and datasets at https://github.com/jdime/scRNAseq_cell_cluster_labeling.
利用单细胞RNA测序(scRNA-seq)数据从复杂细胞混合物中识别细胞类型亚群,包括从标准化到细胞聚类的自动化步骤。然而,给细胞簇分配细胞类型标签通常是手动进行的,这导致记录有限、可重复性低以及词汇不受控制。部分原因是参考细胞类型特征稀缺,且一些方法支持的细胞类型特征有限。在本研究中,我们对代表第一代富集分析(ORA)、第二代方法(GSEA和GSVA)、机器学习工具(CIBERSORT)和基于网络的邻居投票(METANEIGHBOR)的五种方法进行了基准测试,用于从scRNA-seq数据给细胞簇分配细胞类型标签的任务。我们使用了五个scRNA-seq数据集:人类肝脏、11个小鼠组织图谱小鼠组织、两个人类外周血单核细胞数据集以及小鼠视网膜神经元,这些数据集都有可用的参考细胞类型特征。这些数据集涵盖了Drop-seq、10X Chromium和Seq-Well技术,细胞数量从约3700个到约68000个不等。我们的结果表明,总体而言,通过接受者操作特征曲线分析评估,所有五种方法在该任务中表现良好(曲线下平均面积(AUC)=0.91,标准差=0.06),而精确召回分析显示,根据方法和数据集的不同存在很大差异(平均AUC =0.53,标准差=0.24)。我们观察到细胞类型特征中基因数量对性能有影响,特征越小越频繁导致错误结果。GSVA总体上表现最佳,在细胞类型特征二次抽样模拟中更稳健,尽管不同方法在不同数据集上表现良好。METANEIGHBOR和GSVA是最快的方法。与其他方法相比,CIBERSORT和METANEIGHBOR在仅包括预期细胞类型的分析中受影响更大。我们在https://github.com/jdime/scRNAseq_cell_cluster_labeling提供了一个可扩展框架,可用于评估其他方法和数据集。