Department of Bioengineering and Therapeutic Sciences, University of California San Francisco, San Francisco, CA 94158, USA; Department of Biological Sciences, Auburn University, Auburn, AL 36842, USA.
Department of Medicine, University of California San Francisco, San Francisco, CA 94158, USA.
Am J Hum Genet. 2019 Oct 3;105(4):747-762. doi: 10.1016/j.ajhg.2019.08.011. Epub 2019 Sep 19.
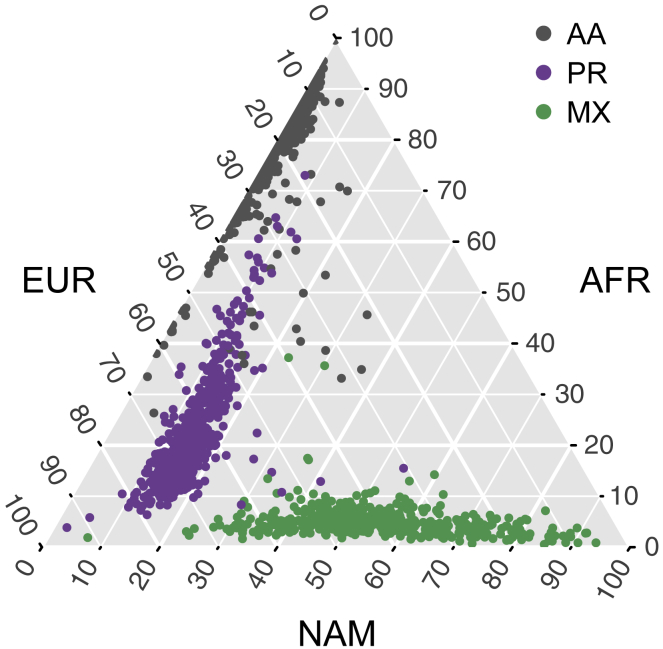
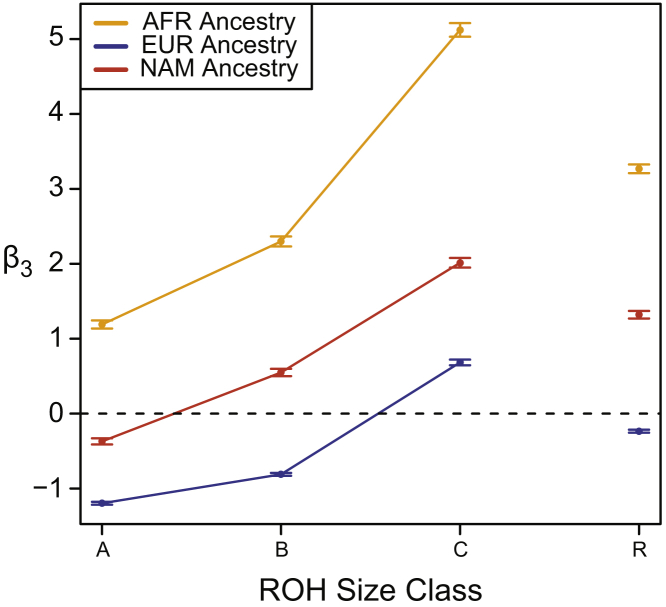
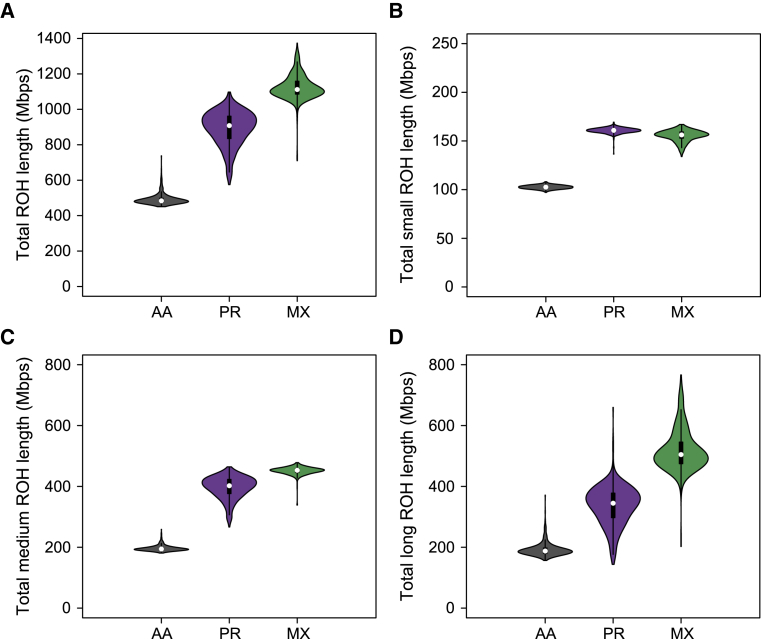
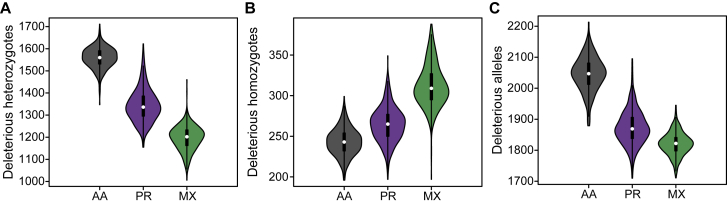
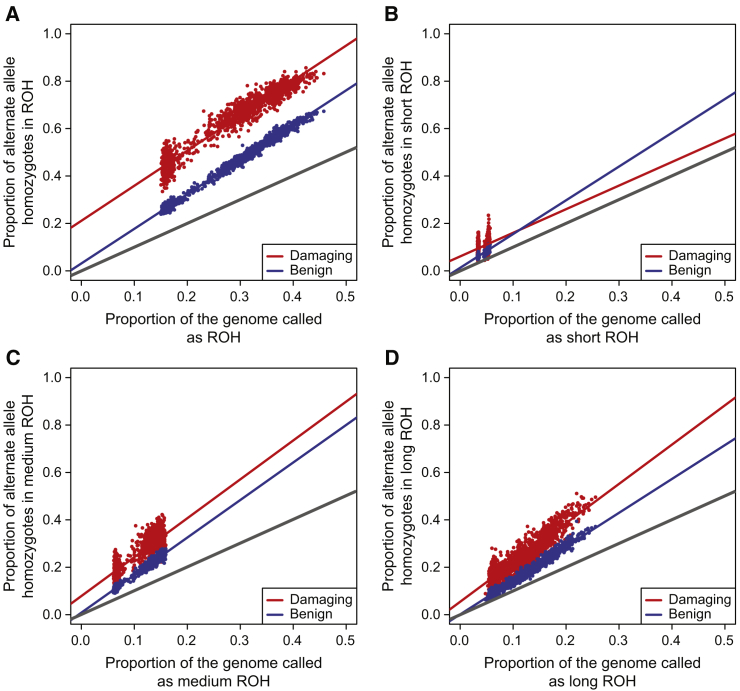
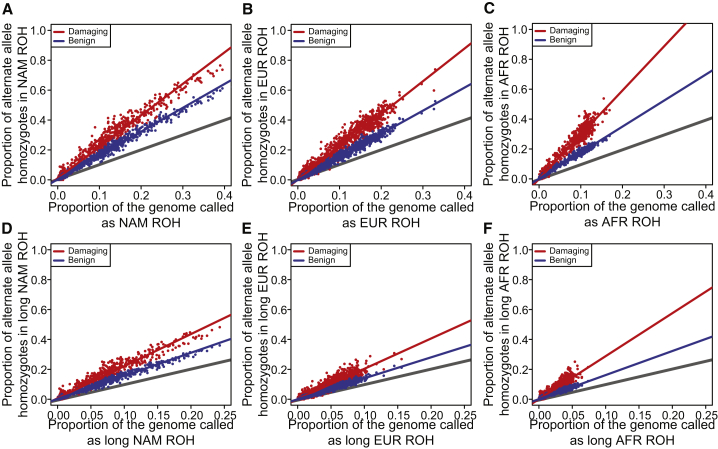
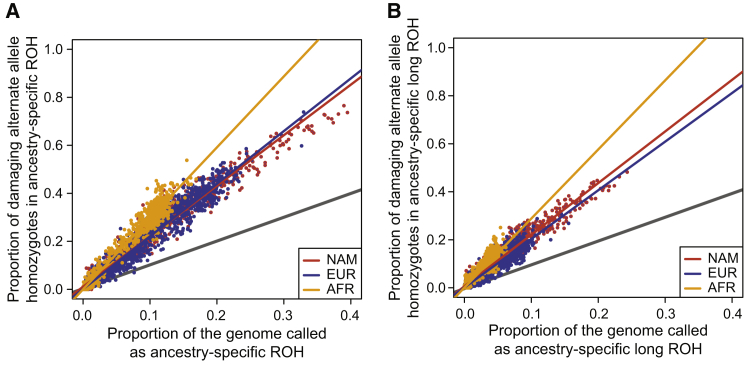
Runs of homozygosity (ROH) are important genomic features that manifest when an individual inherits two haplotypes that are identical by descent. Their length distributions are informative about population history, and their genomic locations are useful for mapping recessive loci contributing to both Mendelian and complex disease risk. We have previously shown that ROH, and especially long ROH that are likely the result of recent parental relatedness, are enriched for homozygous deleterious coding variation in a worldwide sample of outbred individuals. However, the distribution of ROH in admixed populations and their relationship to deleterious homozygous genotypes is understudied. Here we analyze whole-genome sequencing data from 1,441 unrelated individuals from self-identified African American, Puerto Rican, and Mexican American populations. These populations are three-way admixed between European, African, and Native American ancestries and provide an opportunity to study the distribution of deleterious alleles partitioned by local ancestry and ROH. We re-capitulate previous findings that long ROH are enriched for deleterious variation genome-wide. We then partition by local ancestry and show that deleterious homozygotes arise at a higher rate when ROH overlap African ancestry segments than when they overlap European or Native American ancestry segments of the genome. These results suggest that, while ROH on any haplotype background are associated with an inflation of deleterious homozygous variation, African haplotype backgrounds may play a particularly important role in the genetic architecture of complex diseases for admixed individuals, highlighting the need for further study of these populations.
纯合子区域(ROH)是重要的基因组特征,当个体继承两个来自同一祖先的相同单倍型时就会表现出来。它们的长度分布可以提供有关群体历史的信息,其基因组位置对于定位导致孟德尔和复杂疾病风险的隐性基因座非常有用。我们之前已经表明,ROH,尤其是可能是最近父母亲缘关系的结果的长 ROH,在来自世界各地的外群体个体的样本中,与纯合有害编码变异富集。然而,混合人群中的 ROH 分布及其与有害纯合基因型的关系还研究不足。在这里,我们分析了来自 1441 名自我认同的非裔美国人、波多黎各人和墨西哥裔美国人的无关个体的全基因组测序数据。这些人群是欧洲、非洲和美洲原住民三种混合人群,为研究按本地祖源和 ROH 划分的有害等位基因的分布提供了机会。我们重新证实了先前的发现,即长 ROH 在全基因组范围内富含有害变异。然后,我们按本地祖源进行划分,并表明当 ROH 与非洲祖先片段重叠时,有害纯合子的出现率高于与欧洲或美洲原住民祖先片段重叠时的出现率。这些结果表明,虽然任何单倍型背景上的 ROH 都与有害纯合变异的膨胀有关,但非洲单倍型背景可能在混合人群的复杂疾病遗传结构中发挥特别重要的作用,突出了对这些人群进行进一步研究的必要性。