Department of Inorganic Spectroscopy , Max Planck Institute for Chemical Energy Conversion , Stiftstrasse 34-36 , D-45470 , Mülheim an der Ruhr , Germany.
Department of Chemistry , The University of Texas at Austin , Austin , Texas 78712 , United States.
Inorg Chem. 2019 Oct 7;58(19):12918-12932. doi: 10.1021/acs.inorgchem.9b01870. Epub 2019 Sep 25.
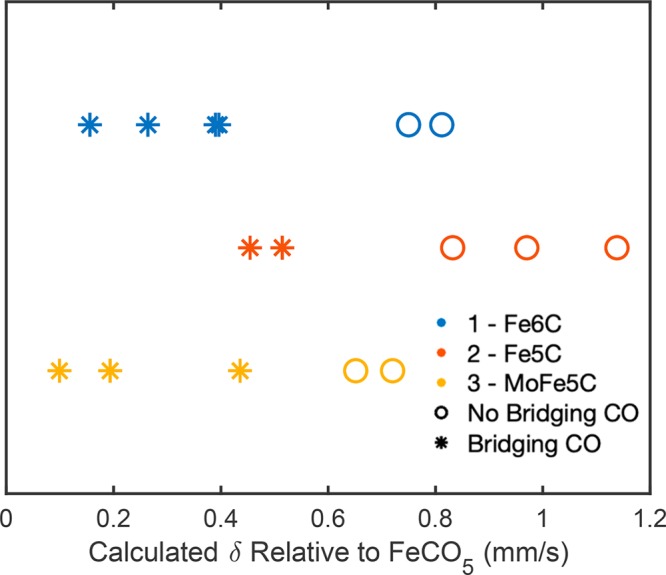
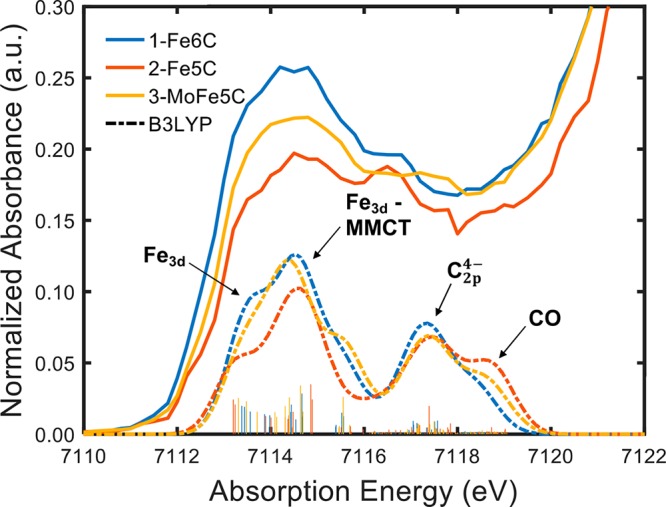
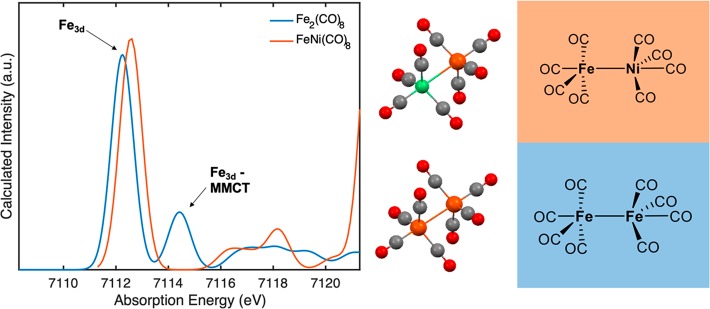
The present study employs a suite of spectroscopic techniques to evaluate the electronic and bonding characteristics of the interstitial carbide in a set of iron-carbonyl-carbide clusters, one of which is substituted with a molybdenum atom. The MC and MC clusters are the dianions (EtN)[(μ-C)(μ-CO)(CO)] (), [K(benzo-18-crown-6)][(μ-C)(μ-CO)(CO)] (), and [K(benzo-18-crown-6)][(μ-C)(μ-CO)(CO)] (). Because and have the same overall cluster charge (2-) but different numbers of iron sites (: 6 sites → : 5 sites), the metal atoms of are formally oxidized compared to those in . Despite this, Mössbauer studies indicate that the iron sites in possess (lower spectroscopic oxidation state) compared with those in . Iron K-edge X-ray absorption and valence-to-core X-ray emission spectroscopy measurements, paired with density functional theory spectral calculations, revealed the presence of significant metal-to-metal and carbide 2p-based character in the filled valence and low-lying unfilled electronic manifolds. In all of the above experiments, the presence of the molybdenum atom in (FeMo) results in somewhat unremarkable spectroscopic properties that are essentially a "hybrid" of (Fe) and (Fe). The overall electronic portrait that emerges illustrates that the central inorganic carbide ligand is essential for distributing charge and maximizing electronic communication throughout the cluster. It is evident that the carbide coordination environment is quite flexible and adaptive: it can drastically modify the covalency of individual Fe-C bonds based on local structural changes and redox manipulation of the clusters. In light of these findings, our data and calculations suggest a potential role for the central carbon atom in FeMoco, which likely performs a similar function in order to maintain cluster integrity through multiple redox and ligand binding events.
本研究采用了一系列光谱技术来评估一组铁-羰基-碳化物簇中间隙碳化物的电子和键合特性,其中一个簇被钼原子取代。MC 和 MC 簇是二阴离子((EtN)[(μ-C)(μ-CO)(CO)] ()、[K(苯并-18-冠-6)][(μ-C)(μ-CO)(CO)] ()和[K(苯并-18-冠-6)][(μ-C)(μ-CO)(CO)] ()。由于和具有相同的总簇电荷(2-),但铁位点的数量不同(:6 个位点→:5 个位点),因此与相比,的金属原子被形式氧化。尽管如此,穆斯堡尔研究表明,与相比,中铁位点具有较低的(较低的光谱氧化态)。铁 K 边 X 射线吸收和价态到核 X 射线发射光谱测量,加上密度泛函理论光谱计算,表明在充满价和低未占据电子简并态中存在显著的金属间和碳化物 2p 基特征。在所有上述实验中,钼原子的存在在(FeMo)中导致了一些不显著的光谱特性,这些特性基本上是(Fe)和(Fe)的“混合”。总体电子图像表明,中心无机碳化物配体对于在整个簇中分配电荷和最大化电子通讯至关重要。显然,碳化物配位环境非常灵活和适应性强:它可以根据局部结构变化和簇的氧化还原操作,极大地改变单个 Fe-C 键的共价性。鉴于这些发现,我们的数据和计算表明中心碳原子在 FeMoco 中可能具有潜在的作用,它可能通过多个氧化还原和配体结合事件来维持簇的完整性,从而发挥类似的功能。