Department of Pharmaceutical Biochemistry, College of Pharmacy, Kyung Hee University, Seoul 02447, Korea.
Department of Life and Nanopharmaceutical Sciences, Graduate School, Kyung Hee University, Seoul 02447, Korea.
Cells. 2019 Sep 27;8(10):1163. doi: 10.3390/cells8101163.
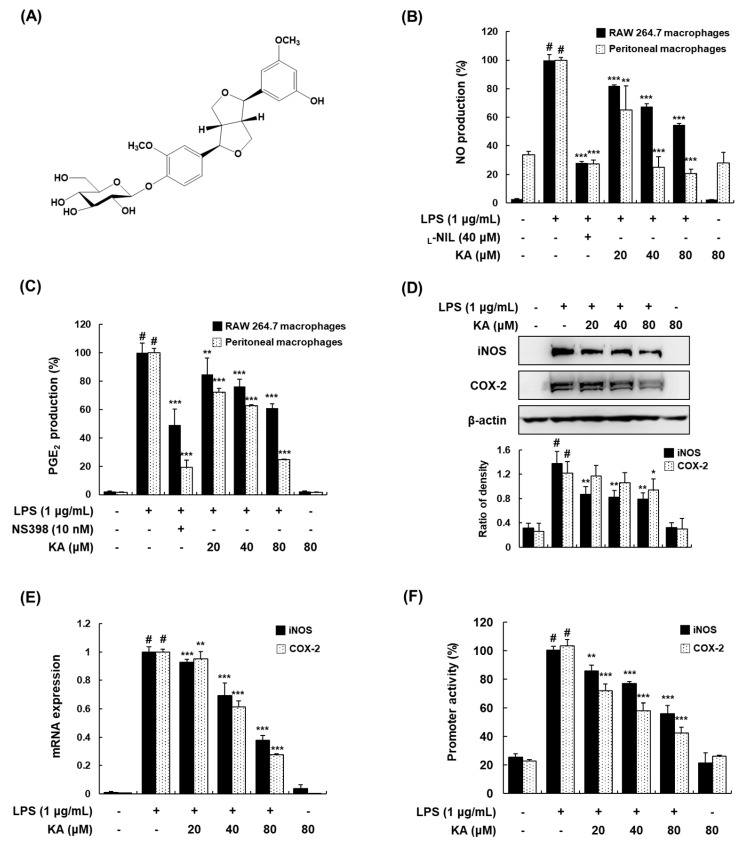
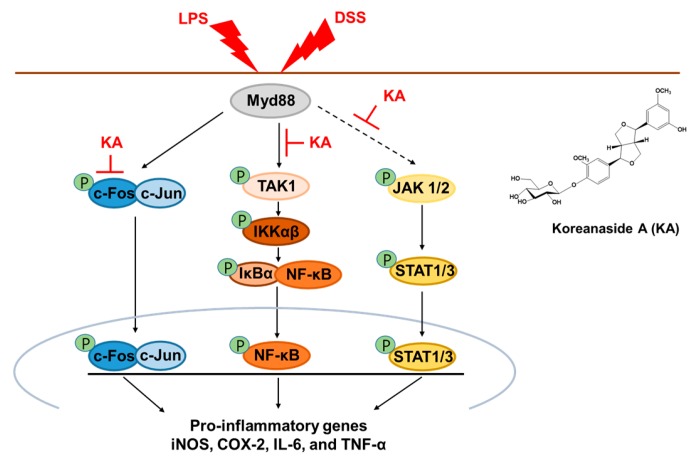
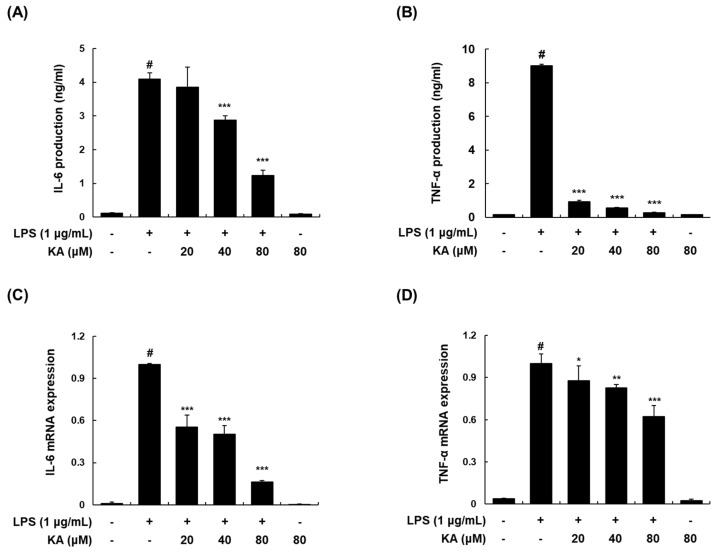
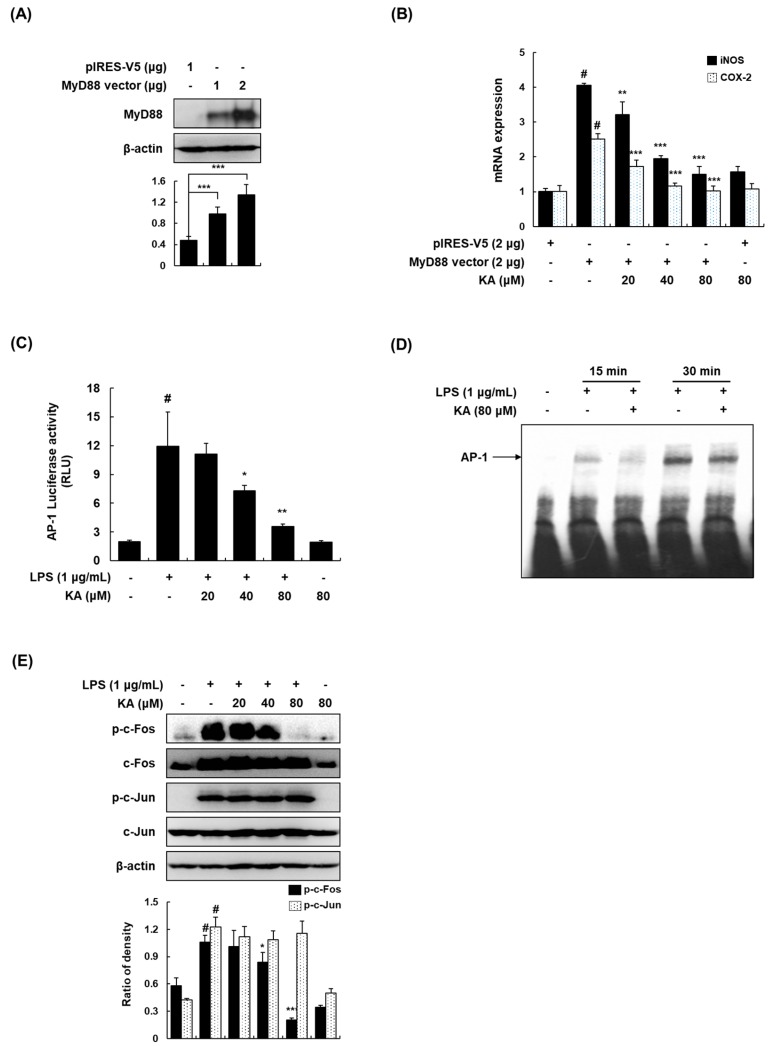
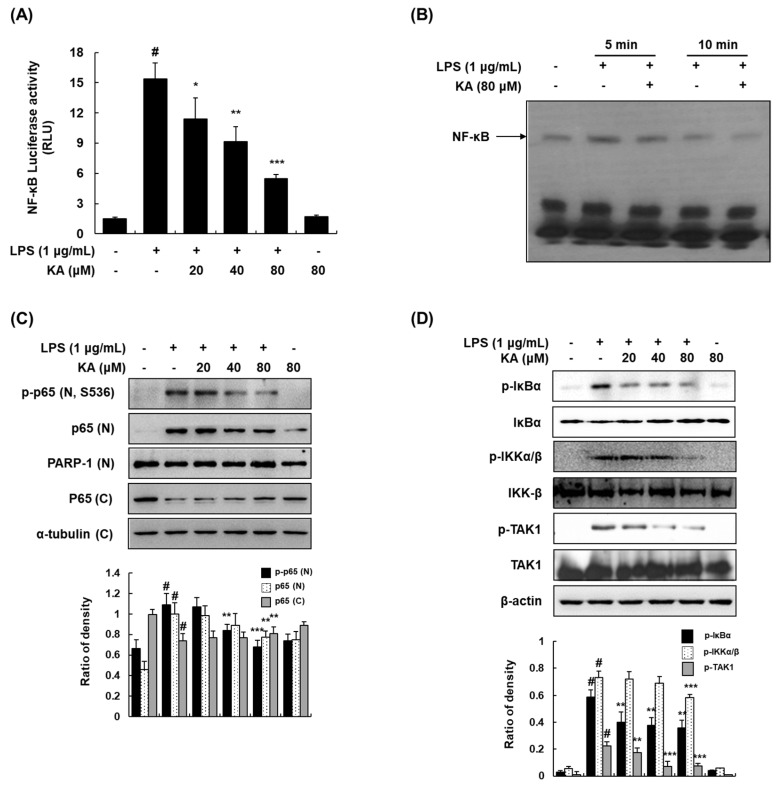
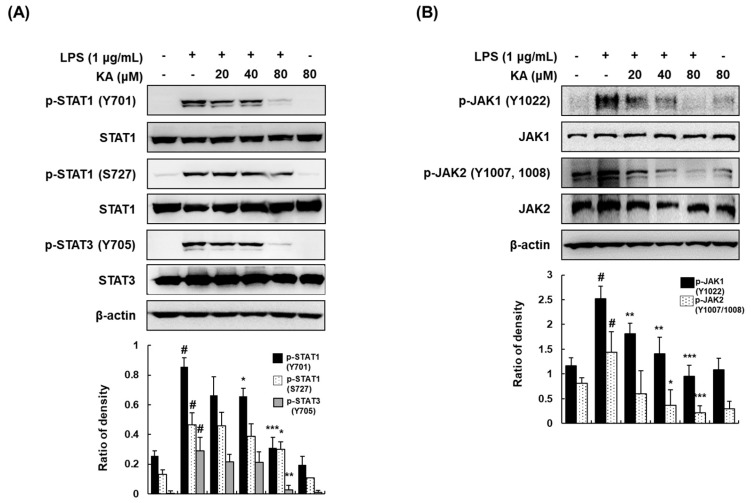
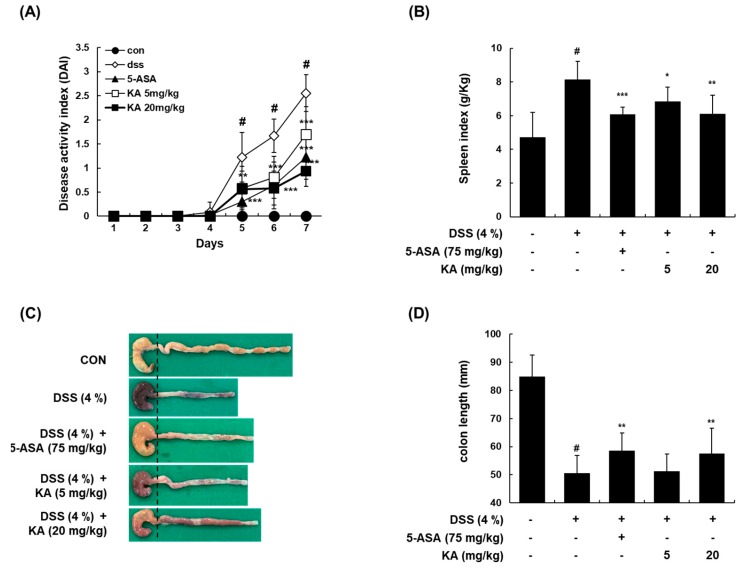
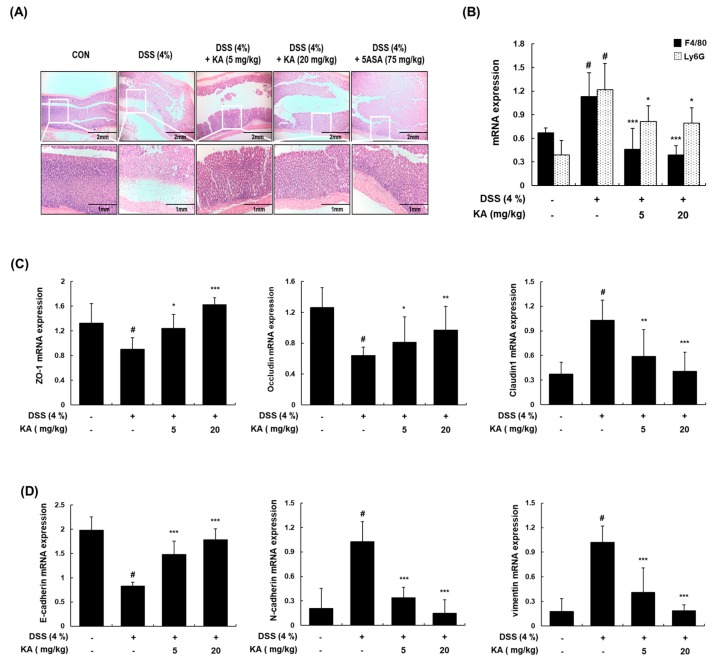
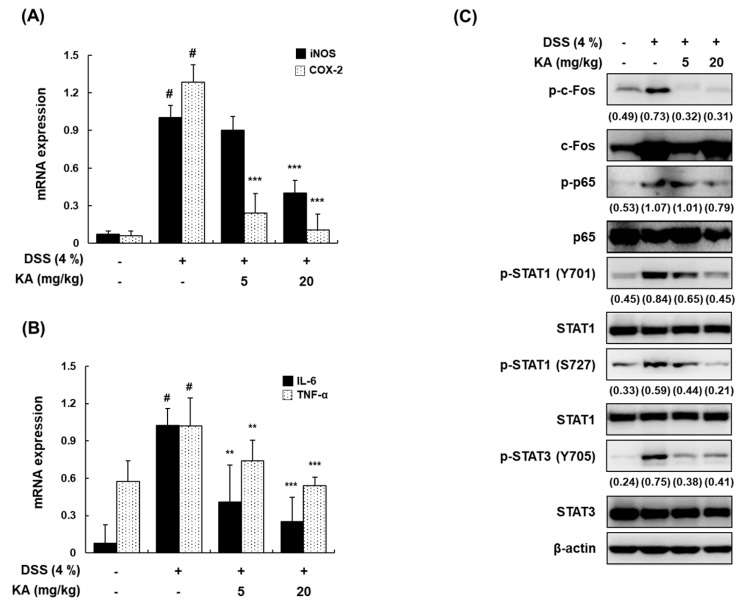
The current treatment options for inflammatory bowel disease (IBD) are unsatisfactory. Therefore, novel and safer therapies are needed. We previously reported that koreanaside A (KA) showed high radical scavenging activity and suppressed vascular cell adhesion molecule 1 (VCAM-1) expression in vascular smooth muscle cells. However, the molecular mechanisms involved in its anti-inflammatory effect have not been reported. KA inhibited pro-inflammatory mediators such as inducible nitric oxide synthase (iNOS), cyclooxygenase-2 (COX-2), nitric oxide (NO), and prostaglandin E (PGE). KA inhibited the production and mRNA expression of interleukin (IL)-6 and tumor necrosis factor-α (TNF-α) induced by LPS. KA downregulated the myeloid differentiation primary response 88 (MyD88)-dependent inflammatory gene expressions in the MyD88-overexpressed cells. KA suppressed the LPS-induced transcriptional and DNA-binding activities of activator protein-1 (AP-1) and nuclear factor-kappa B (NF-κB). KA was found to inhibit the phosphorylation of Janus kinase 1/2 (JAK1/2) and signal transducers and activators of transcription 1/3 (STAT1/3). In DSS-induced colitis mice, KA relieved the symptoms of colitis by suppressing inflammatory cell infiltration, restoring tight junction (TJ)- and epithelial-mesenchymal transition (EMT)-related protein expression, and inactivating AP-1, NF-κB, and STAT1/3. Therefore, KA reduced inflammatory responses by downregulating AP-1, NF-κB, and JAK/STAT signaling in LPS-induced macrophages and DSS-induced colitis mice.
目前治疗炎症性肠病(IBD)的方法并不令人满意。因此,需要寻找新的、更安全的治疗方法。我们之前报道过,高丽当归素 A(KA)表现出很高的自由基清除活性,并抑制血管平滑肌细胞中血管细胞黏附分子 1(VCAM-1)的表达。然而,其抗炎作用的分子机制尚未报道。KA 抑制了促炎介质如诱导型一氧化氮合酶(iNOS)、环氧化酶-2(COX-2)、一氧化氮(NO)和前列腺素 E(PGE)的产生。KA 抑制了 LPS 诱导的白细胞介素(IL)-6 和肿瘤坏死因子-α(TNF-α)的产生和 mRNA 表达。KA 下调了 MyD88 过表达细胞中 MyD88 依赖性炎症基因的表达。KA 抑制了 LPS 诱导的激活蛋白-1(AP-1)和核因子-κB(NF-κB)的转录和 DNA 结合活性。KA 被发现抑制了 Janus 激酶 1/2(JAK1/2)和信号转导和转录激活物 1/3(STAT1/3)的磷酸化。在 DSS 诱导的结肠炎小鼠中,KA 通过抑制炎症细胞浸润、恢复紧密连接(TJ)和上皮-间充质转化(EMT)相关蛋白的表达以及使 AP-1、NF-κB 和 STAT1/3 失活,缓解了结肠炎的症状。因此,KA 通过下调 LPS 诱导的巨噬细胞和 DSS 诱导的结肠炎小鼠中的 AP-1、NF-κB 和 JAK/STAT 信号通路,减轻了炎症反应。