EA2106 BBV, Université de Tours, Tours, 37200, France.
EA3142 GEIHP, Université d'Angers, Université Bretagne-Loire, Angers, 49100, France.
Sci Rep. 2019 Oct 8;9(1):14431. doi: 10.1038/s41598-019-50885-8.
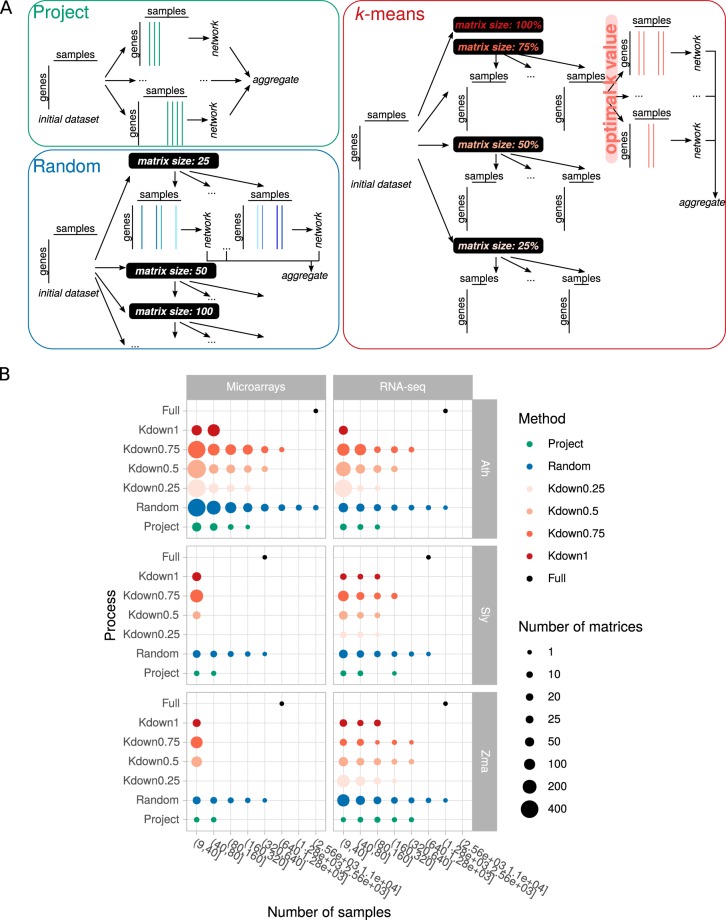
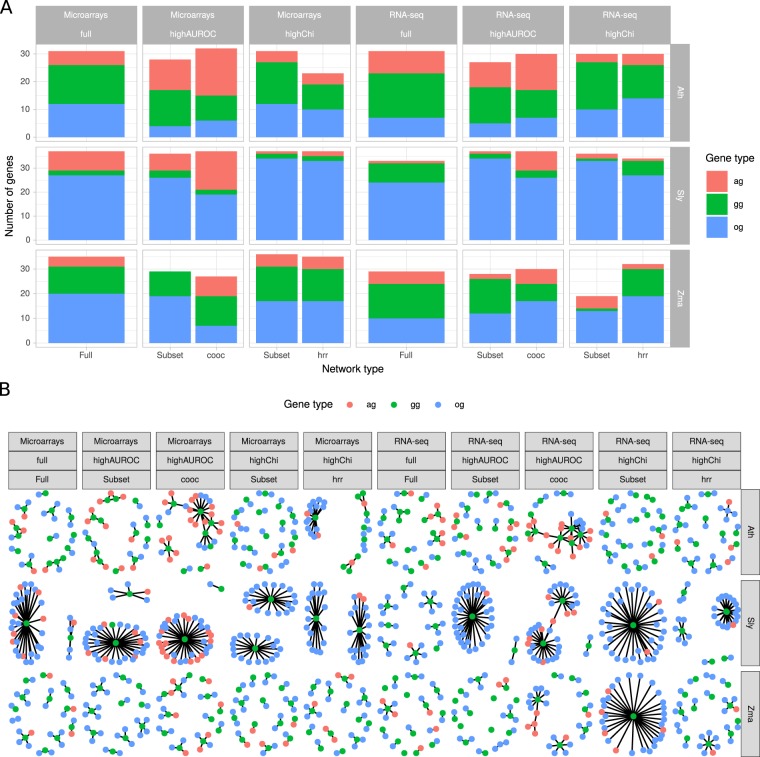
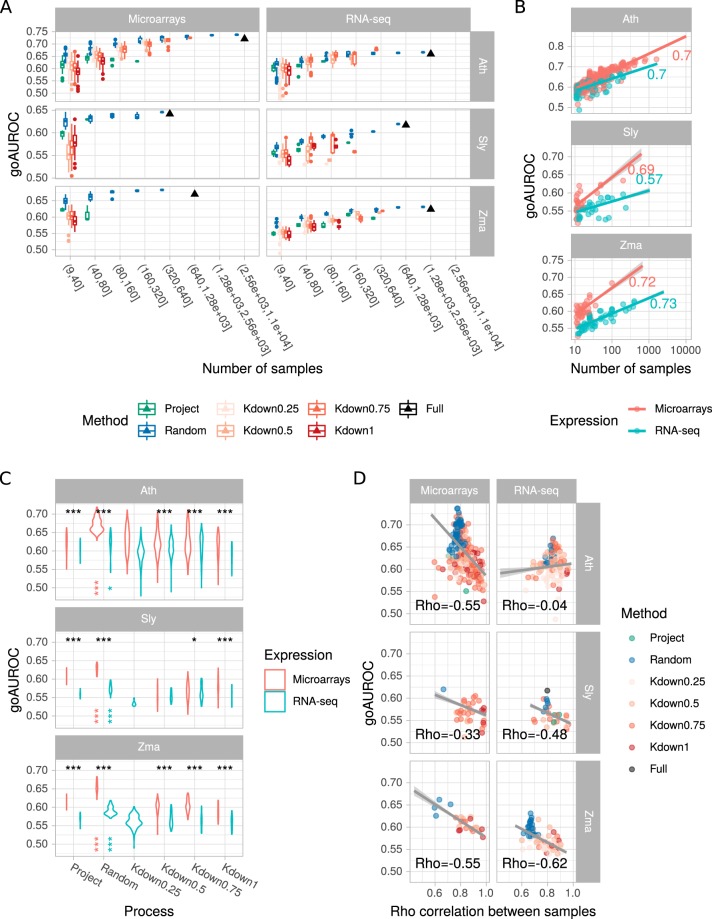
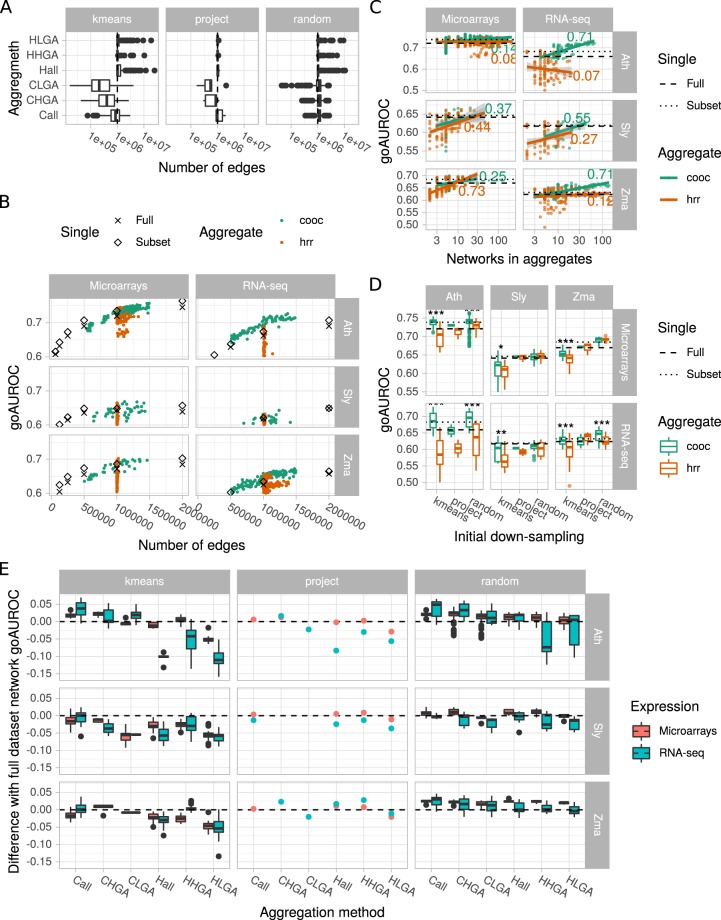
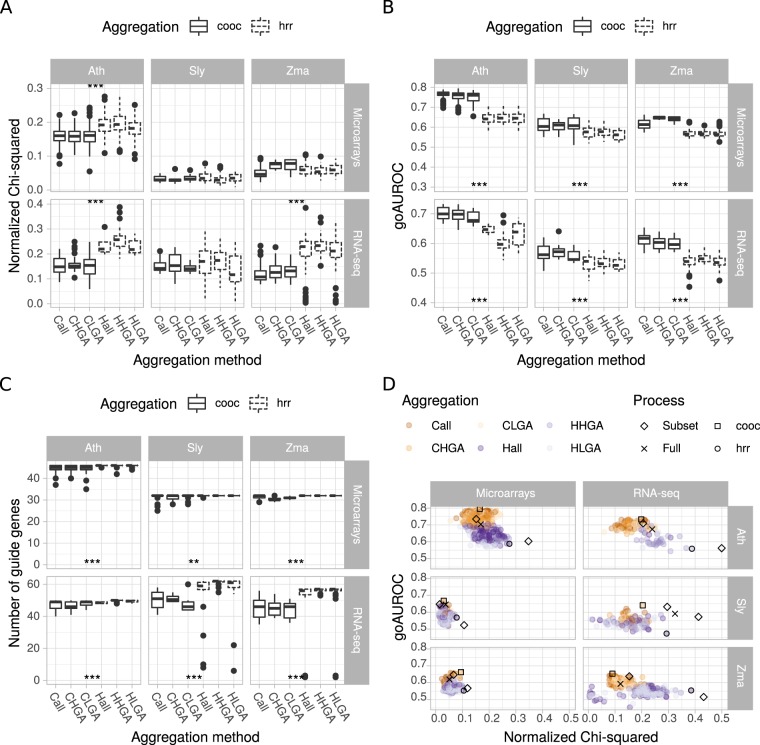
Large-scale gene co-expression networks are an effective methodology to analyze sets of co-expressed genes and discover new gene functions or associations. Distances between genes are estimated according to their expression profiles and are visualized in networks that may be further partitioned to reveal communities of co-expressed genes. Creating expression profiles is now eased by the large amounts of publicly available expression data (microarrays and RNA-seq). Although many distance calculation methods have been intensively compared and reviewed in the past, it is unclear how to proceed when many samples reflecting a wide range of different conditions are available. Should as many samples as possible be integrated into network construction or be partitioned into smaller sets of more related samples? Previous studies have indicated a saturation in network performances to capture known associations once a certain number of samples is included in distance calculations. Here, we examined the influence of sample size on co-expression network construction using microarray and RNA-seq expression data from three plant species. We tested different down-sampling methods and compared network performances in recovering known gene associations to networks obtained from full datasets. We further examined how aggregating networks may help increase this performance by testing six aggregation methods.
大规模基因共表达网络是分析一组共表达基因并发现新基因功能或关联的有效方法。根据基因的表达谱估计基因之间的距离,并将其可视化在网络中,这些网络可以进一步划分以揭示共表达基因的群落。由于大量公开可用的表达数据(微阵列和 RNA-seq),现在创建表达谱变得更加容易。尽管过去已经对许多距离计算方法进行了深入比较和综述,但当有许多反映广泛不同条件的样本可用时,如何进行仍然不清楚。是否应该尽可能多地将样本整合到网络构建中,还是将其划分为更小的、更相关的样本集?先前的研究表明,一旦在距离计算中包含了一定数量的样本,网络性能就会达到捕获已知关联的饱和点。在这里,我们使用来自三个植物物种的微阵列和 RNA-seq 表达数据研究了样本量对共表达网络构建的影响。我们测试了不同的下采样方法,并比较了恢复已知基因关联的网络性能与从完整数据集获得的网络性能。我们进一步研究了通过测试六种聚合方法,聚合网络如何帮助提高这种性能。