Vekris Antonios, Pilalis Eleftherios, Chatziioannou Aristotelis, Petry Klaus G
UMR 1049 and U1029, INSERM, Bordeaux, France.
Metabolic Engineering and Bioinformatics Program, Institute of Chemical Biology, National Hellenic Research Foundation, Athens, Greece.
Front Physiol. 2019 Sep 24;10:1160. doi: 10.3389/fphys.2019.01160. eCollection 2019.
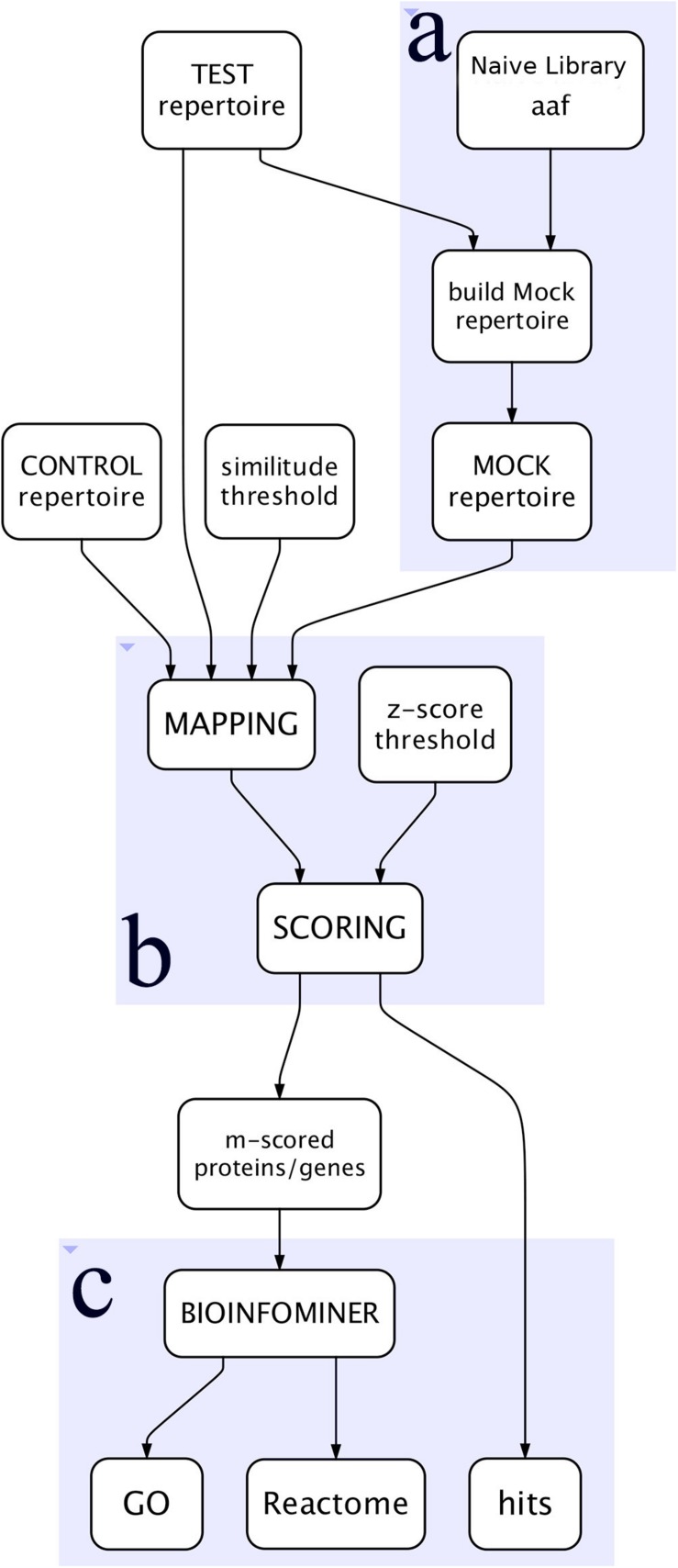
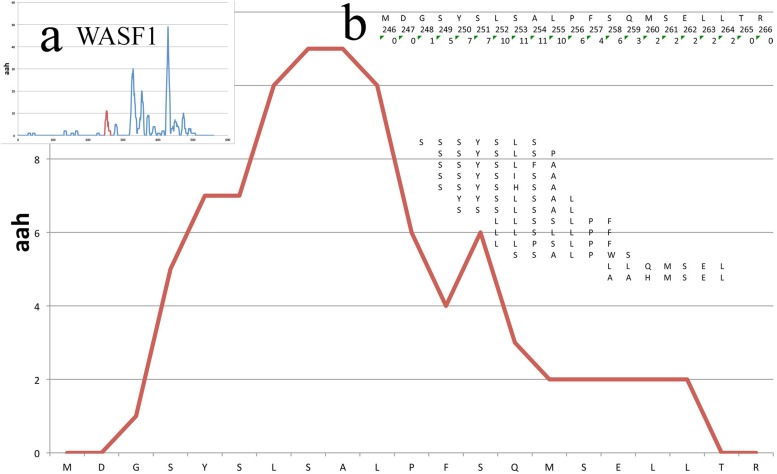
Phage Display is a powerful method for the identification of peptide binding to targets of variable complexities and tissues, from unique molecules to the internal surfaces of vessels of living organisms. Particularly for screenings, the resulting repertoires can be very complex and difficult to study with traditional approaches. Next Generation Sequencing (NGS) opened the possibility to acquire high resolution overviews of such repertoires and thus facilitates the identification of binders of interest. Additionally, the ever-increasing amount of available genome/proteome information became satisfactory regarding the identification of putative mimicked proteins, due to the large scale on which partial sequence homology is assessed. However, the subsequent production of massive data stresses the need for high-performance computational approaches in order to perform standardized and insightful molecular network analysis. Systems-level analysis is essential for efficient resolution of the underlying molecular complexity and the extraction of actionable interpretation, in terms of systemic biological processes and pathways that are systematically perturbed. In this work we introduce PepSimili, an integrated workflow tool, which performs mapping of massive peptide repertoires on whole proteomes and delivers a streamlined, systems-level biological interpretation. The tool employs modules for modeling and filtering of background noise due to random mappings and amplifies the biologically meaningful signal through coupling with BioInfoMiner, a systems interpretation tool that employs graph-theoretic methods for prioritization of systemic processes and corresponding driver genes. The current implementation exploits the Galaxy environment and is available online. A case study using public data is presented, with and without a control selection.
噬菌体展示是一种强大的方法,可用于鉴定与具有不同复杂性的靶标和组织结合的肽,这些靶标和组织范围从独特分子到生物体血管的内表面。特别是在筛选方面,所产生的文库可能非常复杂,难以用传统方法进行研究。下一代测序(NGS)为获取此类文库的高分辨率概况提供了可能性,从而有助于鉴定感兴趣的结合物。此外,由于在大规模上评估了部分序列同源性,越来越多的可用基因组/蛋白质组信息在鉴定推定的模拟蛋白方面变得令人满意。然而,随后产生的大量数据强调了需要高性能的计算方法,以便进行标准化和有洞察力的分子网络分析。系统水平分析对于有效解决潜在的分子复杂性以及从系统生物学过程和途径系统扰动的角度提取可操作的解释至关重要。在这项工作中,我们引入了PepSimili,这是一种集成的工作流程工具,它可以在整个蛋白质组上对大量肽文库进行映射,并提供简化的系统水平生物学解释。该工具采用模块对由于随机映射产生的背景噪声进行建模和过滤,并通过与BioInfoMiner耦合来放大具有生物学意义的信号,BioInfoMiner是一种系统解释工具,它采用图论方法对系统过程和相应的驱动基因进行优先级排序。当前的实现利用了Galaxy环境并且可以在线获取。本文展示了一个使用公共数据的案例研究,包括有和没有对照选择的情况。