Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, Gaustadalléen 21, Oslo, 0318, Norway.
Department of Biostatistics, Harvard T.H. Chan School of Public Health, 677 Huntington Ave, Boston, 02215, USA.
BMC Cancer. 2019 Oct 25;19(1):1003. doi: 10.1186/s12885-019-6235-7.
In biomedical research, network inference algorithms are typically used to infer complex association patterns between biological entities, such as between genes or proteins, using data from a population. This resulting aggregate network, in essence, averages over the networks of those individuals in the population. LIONESS (Linear Interpolation to Obtain Network Estimates for Single Samples) is a method that can be used together with a network inference algorithm to extract networks for individual samples in a population. The method's key characteristic is that, by modeling networks for individual samples in a data set, it can capture network heterogeneity in a population. LIONESS was originally made available as a function within the PANDA (Passing Attributes between Networks for Data Assimilation) regulatory network reconstruction framework. However, the LIONESS algorithm is generalizable and can be used to model single sample networks based on a wide range of network inference algorithms.
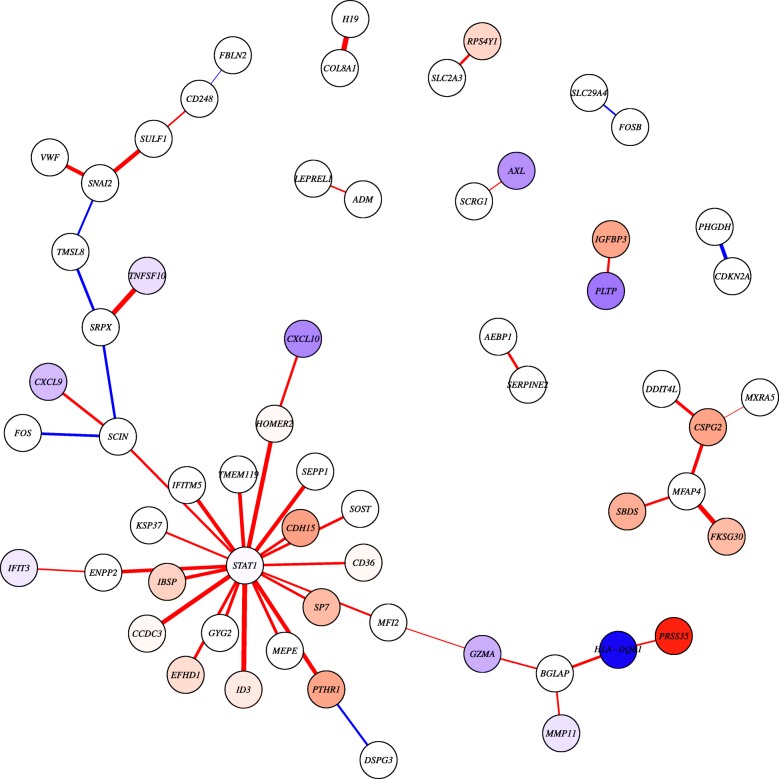
In this software article, we describe lionessR, an R implementation of LIONESS that can be applied to any network inference method in R that outputs a complete, weighted adjacency matrix. As an example, we provide a vignette of an application of lionessR to model single sample networks based on correlated gene expression in a bone cancer dataset. We show how the tool can be used to identify differential patterns of correlation between two groups of patients.
We developed lionessR, an open source R package to model single sample networks. We show how lionessR can be used to inform us on potential precision medicine applications in cancer. The lionessR package is a user-friendly tool to perform such analyses. The package, which includes a vignette describing the application, is freely available at: https://github.com/kuijjerlab/lionessR and at: http://bioconductor.org/packages/lionessR .
在生物医学研究中,网络推断算法通常用于使用来自人群的数据推断生物实体之间(如基因或蛋白质之间)复杂的关联模式。由此产生的综合网络本质上是对人群中个体网络的平均。LIONESS(线性插值以获取单个样本的网络估计值)是一种可以与网络推断算法一起使用的方法,用于从人群中的单个样本中提取网络。该方法的关键特征是,通过对数据集内的个体样本的网络进行建模,可以捕获人群中的网络异质性。LIONESS 最初作为 PANDA(网络之间传递属性以进行数据同化)调控网络重建框架中的一个函数提供。然而,LIONESS 算法具有通用性,可以基于广泛的网络推断算法来对单个样本网络进行建模。
在本文中,我们描述了 lionessR,这是一个基于 R 的 LIONESS 实现,可以应用于任何在 R 中输出完整加权邻接矩阵的网络推断方法。作为一个示例,我们提供了一个基于骨癌数据集的相关基因表达来构建单个样本网络的 lionessR 应用的示例。我们展示了如何使用该工具来识别两组患者之间的相关差异模式。
我们开发了 lionessR,这是一个用于构建单个样本网络的开源 R 包。我们展示了如何使用 lionessR 为癌症中的潜在精准医学应用提供信息。lionessR 包是执行此类分析的用户友好工具。该包包括一个描述应用的示例,可在以下网址获得:https://github.com/kuijjerlab/lionessR 和 http://bioconductor.org/packages/lionessR。