Department of Epidemiology, Biostatistics and Occupational Health, McGill University, Montréal, QC, Canada.
Centre for Clinical Epidemiology, Lady Davis Institute, Jewish General Hospital, Montréal, QC, Canada.
Bioinformatics. 2020 Mar 1;36(6):1840-1847. doi: 10.1093/bioinformatics/btz824.
The human microbiota is the collection of microorganisms colonizing the human body, and plays an integral part in human health. A growing trend in microbiome analysis is to construct a network to estimate the co-occurrence patterns among taxa through precision matrices. Existing methods do not facilitate investigation into how these networks change with respect to covariates.
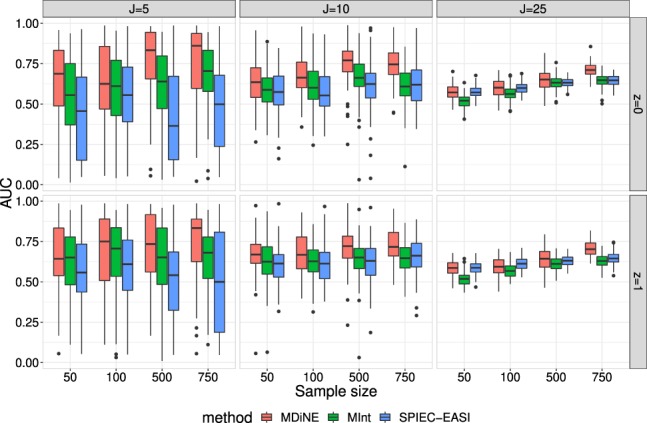
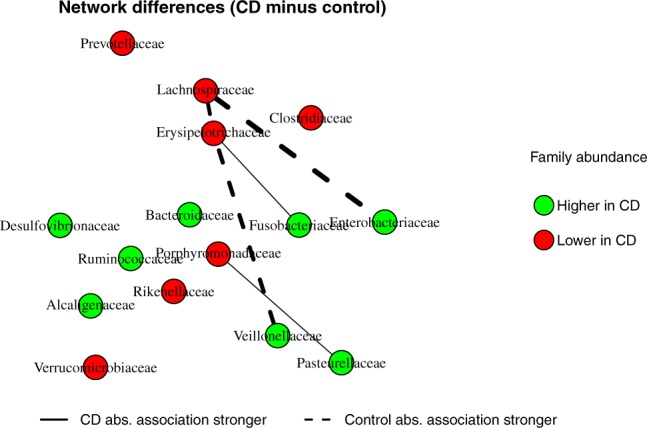
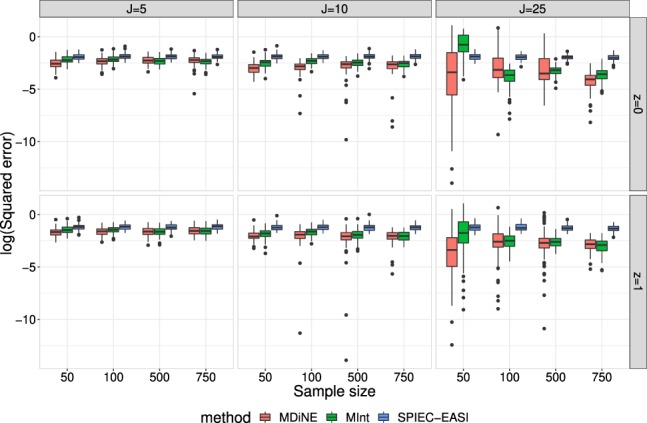
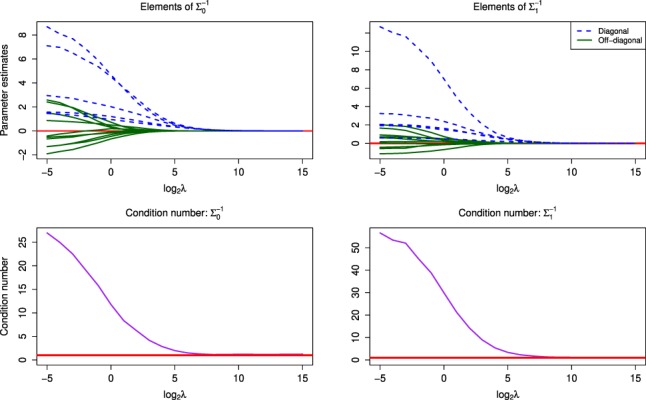
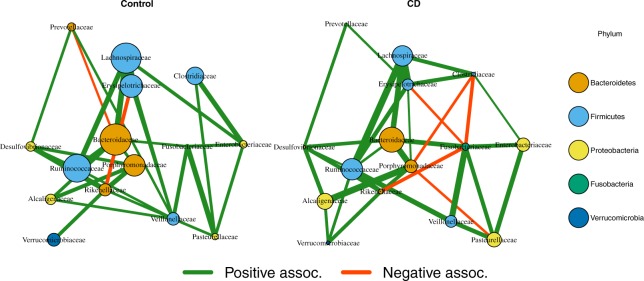
We propose a new model called Microbiome Differential Network Estimation (MDiNE) to estimate network changes with respect to a binary covariate. The counts of individual taxa in the samples are modeled through a multinomial distribution whose probabilities depend on a latent Gaussian random variable. A sparse precision matrix over all the latent terms determines the co-occurrence network among taxa. The model fit is obtained and evaluated using Hamiltonian Monte Carlo methods. The performance of our model is evaluated through an extensive simulation study and is shown to outperform existing methods in terms of estimation of network parameters. We also demonstrate an application of the model to estimate changes in the intestinal microbial network topology with respect to Crohn's disease.
MDiNE is implemented in a freely available R package: https://github.com/kevinmcgregor/mdine.
Supplementary data are available at Bioinformatics online.
人体微生物群是定植于人体的微生物集合,对人类健康起着不可或缺的作用。微生物组分析的一个发展趋势是构建网络,通过精确矩阵来估计分类群之间的共现模式。现有的方法不利于研究这些网络如何随协变量而变化。
我们提出了一种名为微生物组差异网络估计(MDiNE)的新模型,用于估计网络相对于二进制协变量的变化。样本中个体分类群的计数通过一个多项分布建模,其概率取决于一个潜在的高斯随机变量。一个稀疏的全潜在变量的精度矩阵决定了分类群之间的共现网络。使用哈密顿蒙特卡罗方法获得并评估模型拟合。通过广泛的模拟研究评估了我们模型的性能,并表明在网络参数估计方面优于现有方法。我们还展示了该模型在估计克罗恩病相关的肠道微生物网络拓扑变化方面的应用。
MDiNE 是在一个免费提供的 R 包中实现的:https://github.com/kevinmcgregor/mdine。
补充数据可在生物信息学在线获得。