Institute for Mathematics and Computer Science, University of Greifswald, Walther-Rathenau-Str. 47, Greifswald, 17489, Germany.
Center for Functional Genomics of Microbes, University of Greifswald, Felix-Hausdorff-Str. 8, Greifswald, 17489, Germany.
BMC Bioinformatics. 2019 Nov 8;20(1):558. doi: 10.1186/s12859-019-3182-x.
Vast amounts of next generation sequencing RNA data has been deposited in archives, accompanying very diverse original studies. The data is readily available also for other purposes such as genome annotation or transcriptome assembly. However, selecting a subset of available experiments, sequencing runs and reads for this purpose is a nontrivial task and complicated by the inhomogeneity of the data.
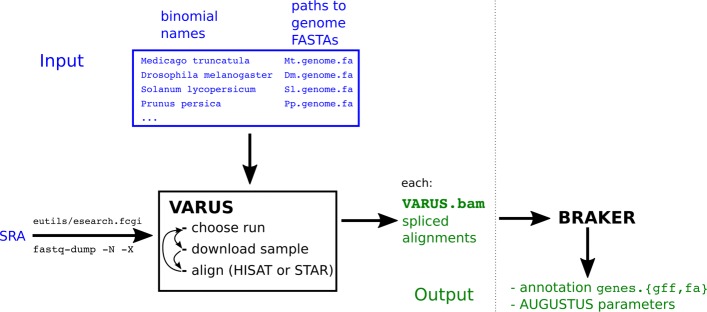
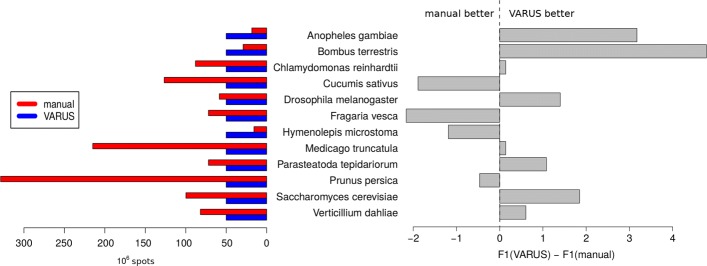
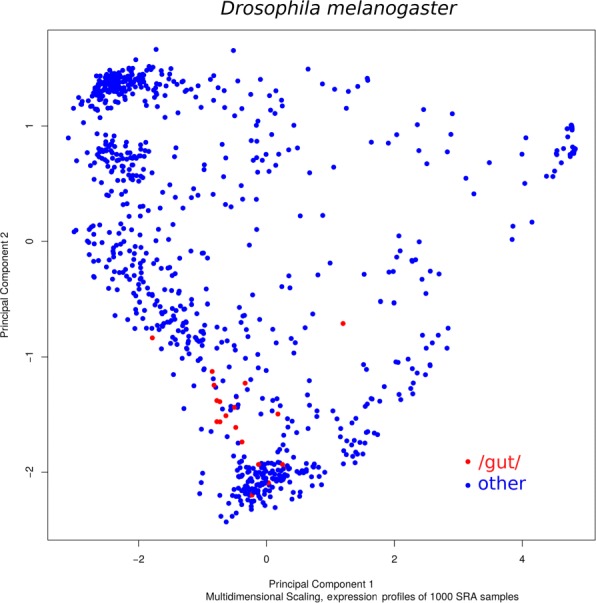
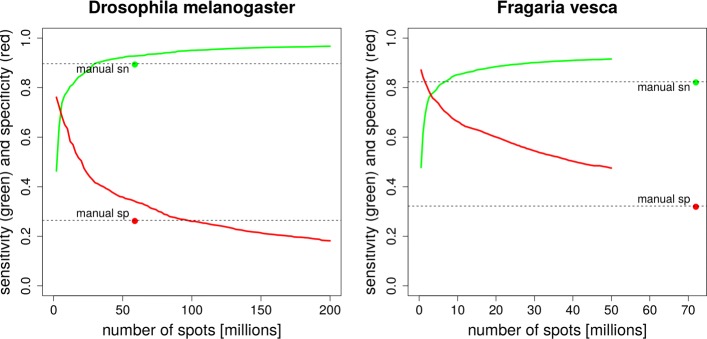
This article presents the software VARUS that selects, downloads and aligns reads from NCBI's Sequence Read Archive, given only the species' binomial name and genome. VARUS automatically chooses runs from among all archived runs to randomly select subsets of reads. The objective of its online algorithm is to cover a large number of transcripts adequately when network bandwidth and computing resources are limited. For most tested species VARUS achieved both a higher sensitivity and specificity with a lower number of downloaded reads than when runs were manually selected. At the example of twelve eukaryotic genomes, we show that RNA-Seq that was sampled with VARUS is well-suited for fully-automatic genome annotation with BRAKER.
With VARUS, genome annotation can be automatized to the extent that not even the selection and quality control of RNA-Seq has to be done manually. This introduces the possibility to have fully automatized genome annotation loops over potentially many species without incurring a loss of accuracy over a manually supervised annotation process.
大量下一代测序 RNA 数据已被存入档案,这些数据伴随了非常多样化的原始研究。这些数据也很容易被用于其他目的,例如基因组注释或转录组组装。然而,为了达到这些目的,选择实验、测序运行和读取的子集是一项非常复杂的任务,并且受到数据不均匀性的影响。
本文介绍了 VARUS 软件,它可以根据物种的二项式名称和基因组,从 NCBI 的序列读取档案中选择、下载和对齐读取。VARUS 可以自动从所有存档运行中选择运行,随机选择读取的子集。其在线算法的目标是在网络带宽和计算资源有限的情况下,充分覆盖大量的转录本。对于大多数测试物种,VARUS 实现了更高的灵敏度和特异性,同时下载的读取数量比手动选择运行时要少。以 12 个真核基因组为例,我们表明,使用 VARUS 采样的 RNA-Seq 非常适合使用 BRAKER 进行全自动基因组注释。
使用 VARUS,基因组注释可以实现自动化,甚至不需要手动进行 RNA-Seq 的选择和质量控制。这就引入了一种可能性,即在不需要人为监督注释过程的准确性损失的情况下,对可能许多物种进行全自动基因组注释循环。