Theoretical Sciences Unit, Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore, India.
PLoS One. 2020 Jan 10;15(1):e0227621. doi: 10.1371/journal.pone.0227621. eCollection 2020.
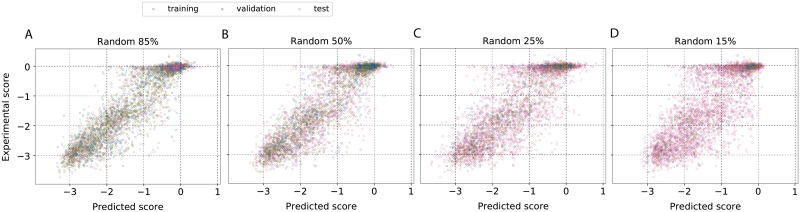
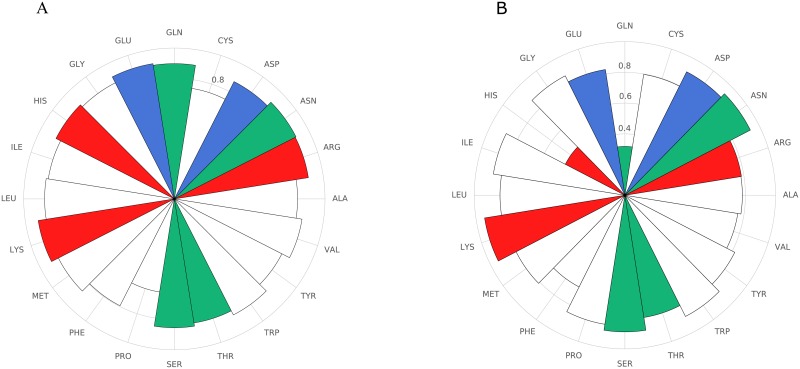
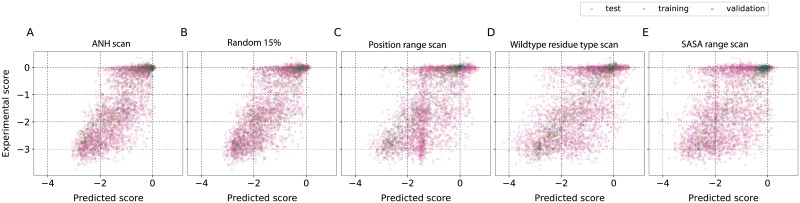
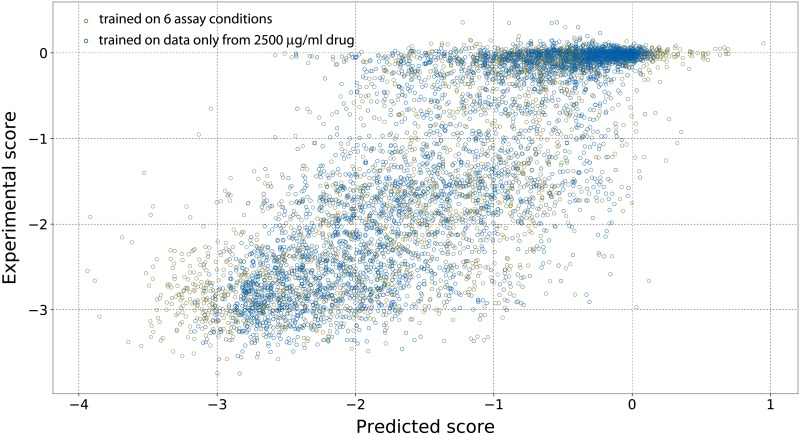
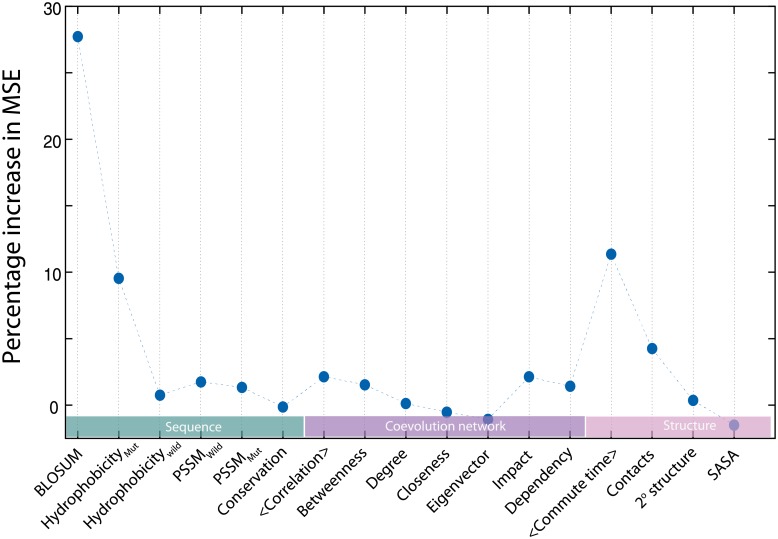
Performing a complete deep mutational scan with all single point mutations may not be practical, and may not even be required, especially if predictive computational models can be developed. Computational models are however naive to cellular response in the myriads of assay-conditions. In a realistic paradigm of assay context-aware predictive hybrid models that combine minimal experimental data from deep mutational scans with structure, sequence information and computational models, we define and evaluate different strategies for choosing this minimal set. We evaluated the trivial strategy of a systematic reduction in the number of mutational studies from 85% to 15%, along with several others about the choice of the types of mutations such as random versus site-directed with the same 15% data completeness. Interestingly, the predictive capabilities by training on a random set of mutations and using a systematic substitution of all amino acids to alanine, asparagine and histidine (ANH) were comparable. Another strategy we explored, augmenting the training data with measurements of the same mutants at multiple assay conditions, did not improve the prediction quality. For the six proteins we analyzed, the bin-wise error in prediction is optimal when 50-100 mutations per bin are used in training the computational model, suggesting that good prediction quality may be achieved with a library of 500-1000 mutations.
进行全面的单点突变深度突变扫描可能不切实际,甚至可能不需要,特别是如果可以开发出预测性计算模型的话。然而,计算模型对于无数种检测条件下的细胞反应来说是幼稚的。在一个将深度突变扫描的最小实验数据与结构、序列信息和计算模型相结合的具有检测上下文意识的预测性混合模型的现实范例中,我们定义并评估了选择这个最小集合的不同策略。我们评估了一种从 85%到 15%的系统减少突变研究数量的简单策略,以及其他几种关于选择突变类型的策略,例如随机突变与相同数据完整度的定点突变。有趣的是,通过在随机突变集上进行训练,并使用系统取代所有的丙氨酸、天冬酰胺和组氨酸(ANH)的方法进行训练,其预测能力是相当的。我们还探索了另一种策略,即在多个检测条件下增加对相同突变体的测量来扩充训练数据,但这并没有提高预测质量。对于我们分析的六个蛋白质,当每个 bin 中使用 50-100 个突变进行计算模型训练时,预测的 bin 误差是最优的,这表明使用 500-1000 个突变的文库可能可以实现良好的预测质量。