Vedithi Sundeep Chaitanya, Malhotra Sony, Skwark Marcin J, Munir Asma, Acebrón-García-De-Eulate Marta, Waman Vaishali P, Alsulami Ali, Ascher David B, Blundell Tom L
Department of Biochemistry, University of Cambridge, Tennis Court Rd., CB2 1GA, UK.
Department of Biological Sciences, Institute of Structural and Molecular Biology, Birkbeck College, University of London, Bloomsbury, London WC1E 7HX, United Kingdom.
Comput Struct Biotechnol J. 2020 Nov 19;18:3692-3704. doi: 10.1016/j.csbj.2020.11.013. eCollection 2020.
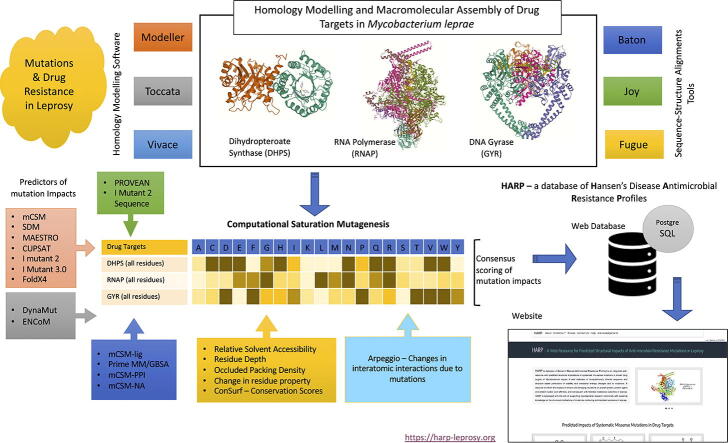
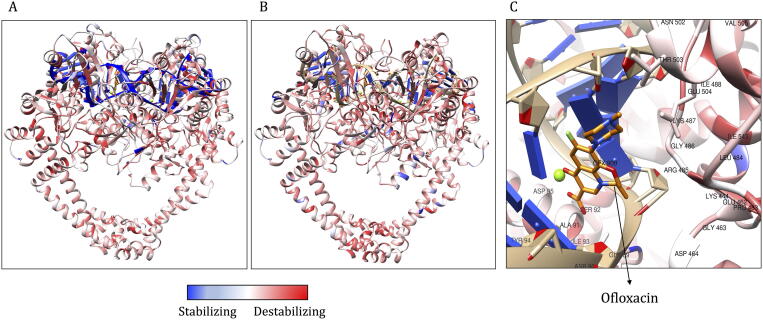
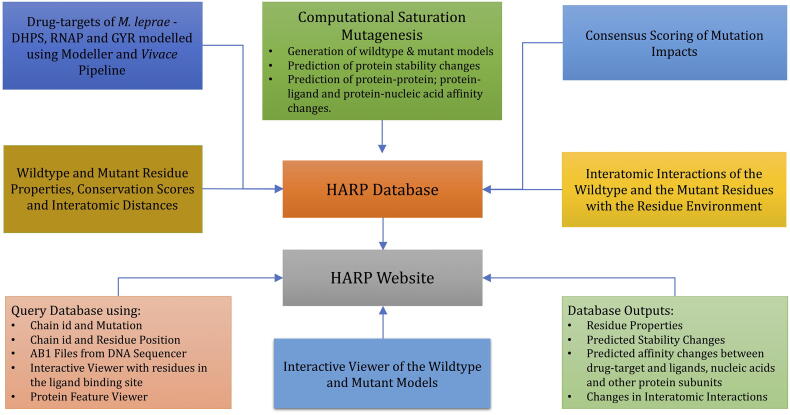
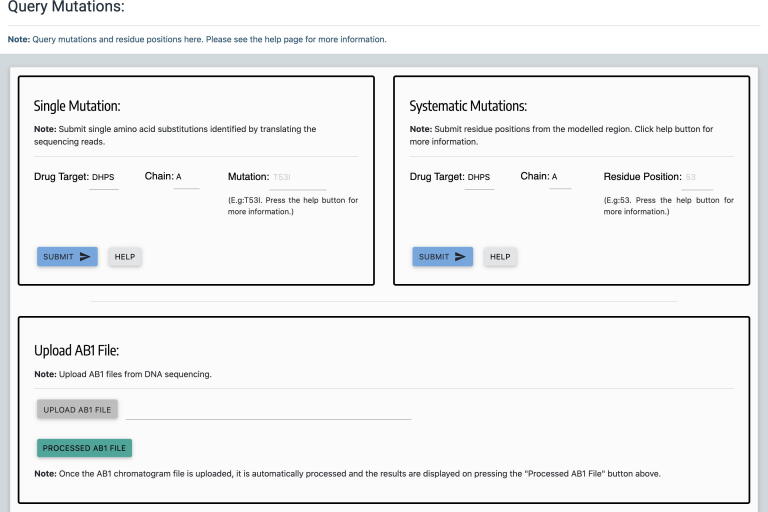
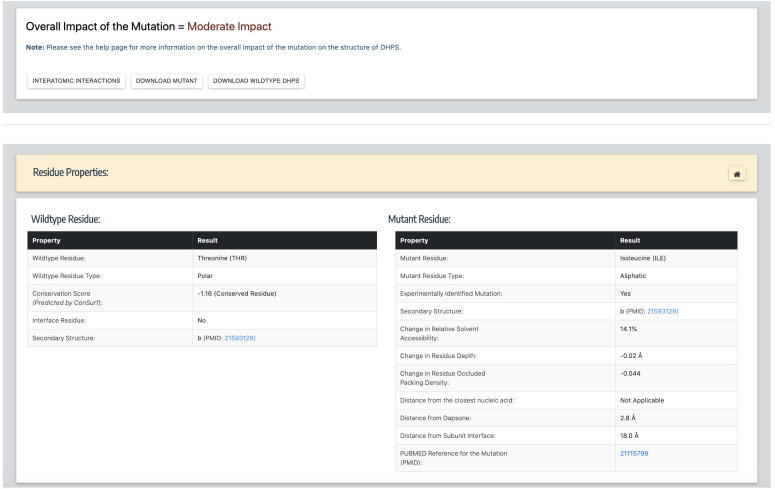
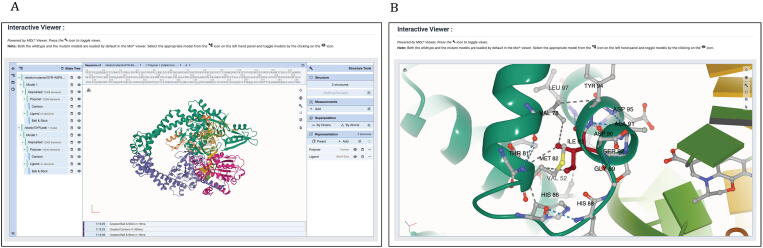
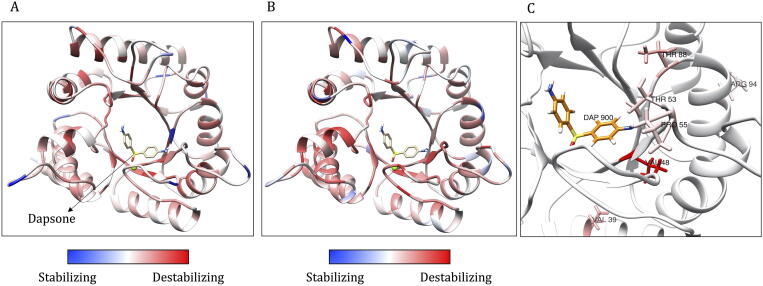
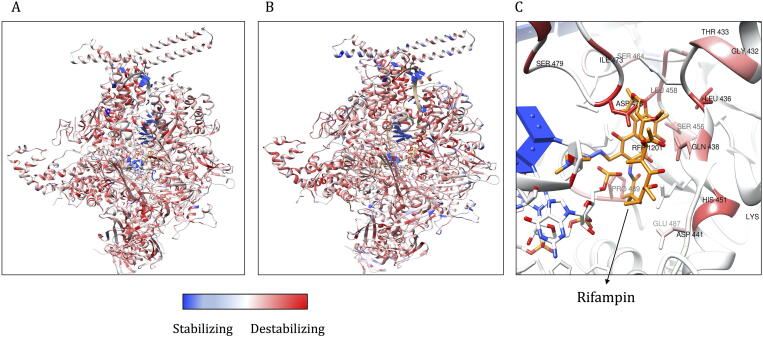
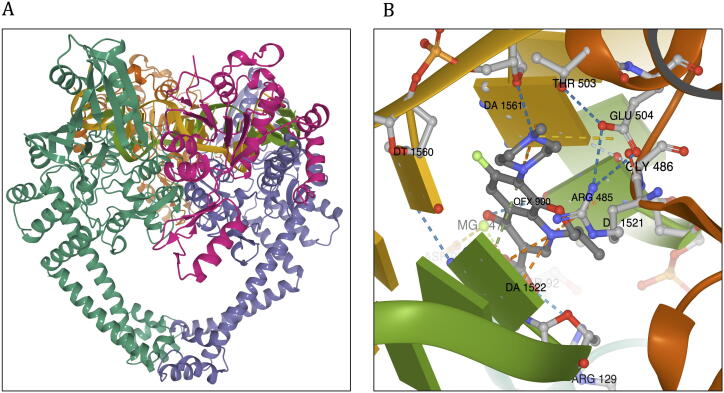
Computational Saturation Mutagenesis is an approach that employs systematic mutagenesis of each amino acid residue in the protein to all other amino acid types, and predicts changes in thermodynamic stability and affinity to the other subunits/protein counterparts, ligands and nucleic acid molecules. The data thus generated are useful in understanding the functional consequences of mutations in antimicrobial resistance phenotypes. In this study, we applied computational saturation mutagenesis to three important drug-targets in for the drugs dapsone, rifampin and ofloxacin namely Dihydropteroate Synthase (DHPS), RNA Polymerase (RNAP) and DNA Gyrase (GYR), respectively. causes leprosy and is an obligate intracellular bacillus with limited protein structural information associating mutations with phenotypic resistance outcomes in leprosy. Experimentally solved structures of DHPS, RNAP and GYR of are not available in the Protein Data Bank, therefore, we modelled the structures of these proteins using template-based comparative modelling and introduced systematic mutations in each model generating 80,902 mutations and mutant structures for all the three proteins. Impacts of mutations on stability and protein-subunit, protein-ligand and protein-nucleic acid affinities were computed using various in-house developed and other published protein stability and affinity prediction software. A consensus impact was estimated for each mutation using qualitative scoring metrics for physicochemical properties and by a categorical grouping of stability and affinity predictions. We developed a web database named HARP (a database of ansen's Disease ntimicrobial esistance rofiles), which is accessible at the URL - and provides the details to each of these predictions.
计算饱和诱变是一种对蛋白质中的每个氨基酸残基进行系统诱变,使其变为所有其他氨基酸类型,并预测其对其他亚基/蛋白质对应物、配体和核酸分子的热力学稳定性和亲和力变化的方法。由此产生的数据有助于理解抗菌耐药表型中突变的功能后果。在本研究中,我们将计算饱和诱变分别应用于氨苯砜、利福平及氧氟沙星三种重要药物的靶点,即二氢蝶酸合酶(DHPS)、RNA聚合酶(RNAP)和DNA促旋酶(GYR)。麻风杆菌会引发麻风病,是一种胞内专性菌,关于其突变与麻风病表型耐药结果相关的蛋白质结构信息有限。蛋白质数据库中没有麻风杆菌DHPS、RNAP和GYR的实验解析结构,因此,我们使用基于模板的比较建模方法对这些蛋白质的结构进行了建模,并在每个模型中引入了系统突变,为这三种蛋白质生成了80902个突变和突变结构。使用各种内部开发的以及其他已发表的蛋白质稳定性和亲和力预测软件,计算了突变对稳定性以及蛋白质 - 亚基、蛋白质 - 配体和蛋白质 - 核酸亲和力的影响。使用物理化学性质的定性评分指标以及稳定性和亲和力预测的分类分组,对每个突变的综合影响进行了估计。我们开发了一个名为HARP(麻风病抗菌耐药谱数据库)的网络数据库,可通过网址 - 访问,该数据库提供了这些预测的详细信息。