Chang Zhiqiang, Miao Xiuxiu, Zhao Wenyuan
College of Bioinformatics Science and Technology, Harbin Medical University, Harbin, China.
Front Genet. 2020 Jan 9;10:1310. doi: 10.3389/fgene.2019.01310. eCollection 2019.
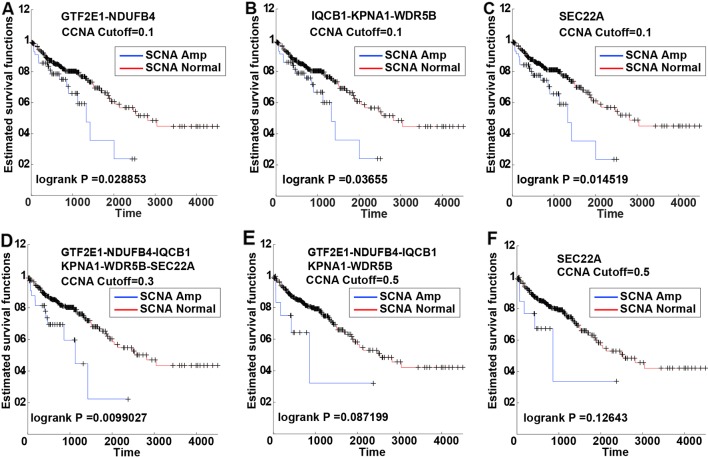
Several studies have already identified the prognostic markers in colorectal cancer (CRC) based on somatic copy number alteration (SCNA). However, very little information is available regarding their value as a prognostic marker. Gene dosage effect is one important mechanism of copy number and dosage-sensitive genes are more likely to behave like driver genes. In this work, we propose a new pipeline to identify the dosage-sensitive prognostic genes in CRC. The RNAseq data, the somatic copy number of CRC from TCGA were assayed to screen out the SCNAs. Wilcoxon rank-sum test was used to identify the differentially expressed genes in alteration samples with |SCNA| > 0.3. Cox-regression was used to find the candidate prognostic genes. An iterative algorithm was built to identify the stable prognostic genes. Finally, the Pearson correlation coefficient was calculated between gene expression and SCNA as the dosage effect score. The cell line data from CCLE was used to test the consistency of the dosage effect. The differential co-expression network was built to discover their function in CRC. A total of six amplified genes (NDUFB4, WDR5B, IQCB1, KPNA1, GTF2E1, and SEC22A) were found to be associated with poor prognosis. They demonstrate a stable prognostic classification in more than 50% threshold of SCNA. The average dosage effect score was 0.5918 ± 0.066, 0.5978 ± 0.082 in TCGA and CCLE, respectively. They also show great stability in different data sets. In the differential co-expression network, these six genes have the top degree and are connected to the driver and tumor suppressor genes. Function enrichment analysis revealed that gene NDUFB4 and GTF2E1 affect cancer-related functions such as transmembrane transport and transformation factors. In conclusion, the pipeline for identifying the prognostic dosage-sensitive genes in CRC was proved to be stable and reliable.
已有多项研究基于体细胞拷贝数改变(SCNA)确定了结直肠癌(CRC)的预后标志物。然而,关于它们作为预后标志物的价值,相关信息却非常少。基因剂量效应是拷贝数的一个重要机制,剂量敏感基因更有可能表现得像驱动基因。在这项研究中,我们提出了一种新的流程来鉴定CRC中剂量敏感的预后基因。对来自TCGA的CRC的RNAseq数据和体细胞拷贝数进行分析,以筛选出SCNA。使用Wilcoxon秩和检验来鉴定|SCNA|>0.3的改变样本中的差异表达基因。使用Cox回归来寻找候选预后基因。构建了一种迭代算法来鉴定稳定的预后基因。最后,计算基因表达与SCNA之间的Pearson相关系数作为剂量效应评分。使用来自CCLE的细胞系数据来测试剂量效应的一致性。构建差异共表达网络以发现它们在CRC中的功能。总共发现六个扩增基因(NDUFB4、WDR5B、IQCB1、KPNA1、GTF2E1和SEC22A)与不良预后相关。它们在超过50%的SCNA阈值时表现出稳定的预后分类。在TCGA和CCLE中,平均剂量效应评分分别为0.5918±0.066和0.5978±0.082。它们在不同数据集中也表现出很高的稳定性。在差异共表达网络中,这六个基因具有最高的度数,并与驱动基因和肿瘤抑制基因相连。功能富集分析表明,基因NDUFB4和GTF2E1影响与癌症相关的功能,如跨膜运输和转化因子。总之,用于鉴定CRC中预后剂量敏感基因的流程被证明是稳定可靠的。