MRC Human Genetics Unit, Institute of Genetics and Molecular Medicine, The University of Edinburgh, Edinburgh, EH4 2XU, UK.
MRC Centre for Regenerative Medicine, Institute for Regeneration and Repair, The University of Edinburgh, Edinburgh, EH16 4UU, UK.
Bioinformatics. 2020 May 1;36(10):2980-2985. doi: 10.1093/bioinformatics/btaa073.
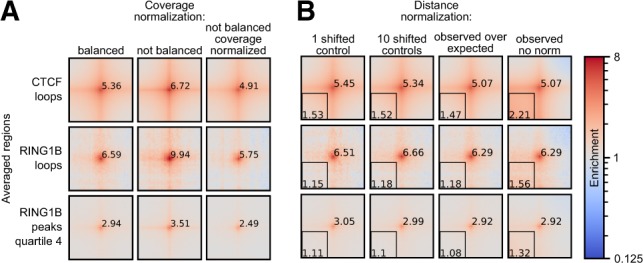
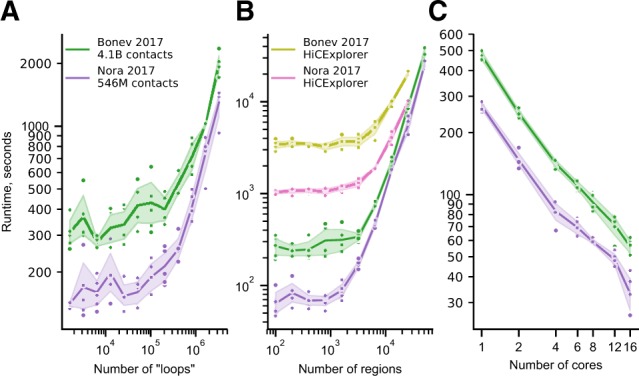
Hi-C is currently the method of choice to investigate the global 3D organization of the genome. A major limitation of Hi-C is the sequencing depth required to robustly detect loops in the data. A popular approach used to mitigate this issue, even in single-cell Hi-C data, is genome-wide averaging (piling-up) of peaks, or other features, annotated in high-resolution datasets, to measure their prominence in less deeply sequenced data. However, current tools do not provide a computationally efficient and versatile implementation of this approach.
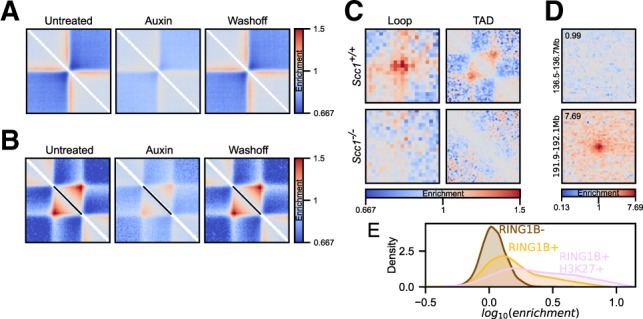
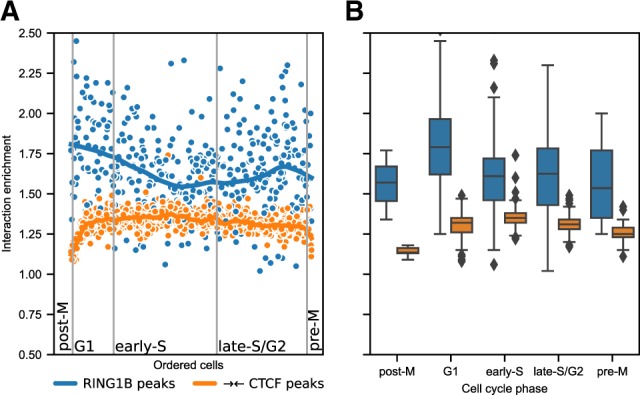
Here, we describe coolpup.py-a versatile tool to perform pile-up analysis on Hi-C data. We demonstrate its utility by replicating previously published findings regarding the role of cohesin and CTCF in 3D genome organization, as well as discovering novel details of Polycomb-driven interactions. We also present a novel variation of the pile-up approach that can aid the statistical analysis of looping interactions. We anticipate that coolpup.py will aid in Hi-C data analysis by allowing easy to use, versatile and efficient generation of pile-ups.
Coolpup.py is cross-platform, open-source and free (MIT licensed) software. Source code is available from https://github.com/Phlya/coolpuppy and it can be installed from the Python Packaging Index.
Hi-C 目前是研究基因组全局 3D 结构的首选方法。Hi-C 的一个主要限制是需要足够的测序深度才能在数据中稳健地检测环。一种流行的方法,即使在单细胞 Hi-C 数据中,也用于减轻这个问题,即在高分辨率数据集中标注的峰或其他特征进行全基因组平均(堆积),以测量它们在测序深度较低的数据中的显著程度。然而,当前的工具并没有提供这种方法的计算效率和多功能性的实现。
在这里,我们描述了 coolpup.py——一种用于在 Hi-C 数据上执行堆积分析的多功能工具。我们通过复制先前关于黏连蛋白和 CTCF 在 3D 基因组组织中的作用的已发表发现,以及发现 Polycomb 驱动相互作用的新细节,证明了其有用性。我们还提出了堆积方法的一种新变体,它可以帮助对环互作用进行统计分析。我们预计 coolpup.py 将通过允许易于使用、多功能和高效的堆积生成来帮助 Hi-C 数据分析。
coolpup.py 是跨平台的、开源的和免费的(MIT 许可证)软件。源代码可从 https://github.com/Phlya/coolpuppy 获得,并可从 Python 包索引中安装。