Taye Zelalem M, Helgason Bobbi L, Bell Jennifer K, Norris Charlotte E, Vail Sally, Robinson Stephen J, Parkin Isobel A P, Arcand Melissa, Mamet Steven, Links Matthew G, Dowhy Tanner, Siciliano Steven, Lamb Eric G
Department of Plant Sciences, College of Agriculture and Bioresources, University of Saskatchewan, Saskatoon, SK, Canada.
Department of Soil Science, College of Agriculture and Bioresources, University of Saskatchewan, Saskatoon, SK, Canada.
Front Microbiol. 2020 Jan 15;10:3007. doi: 10.3389/fmicb.2019.03007. eCollection 2019.
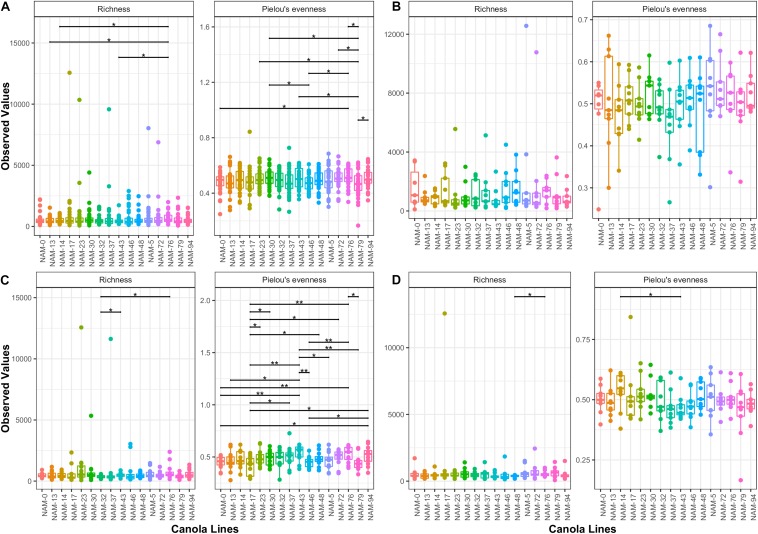
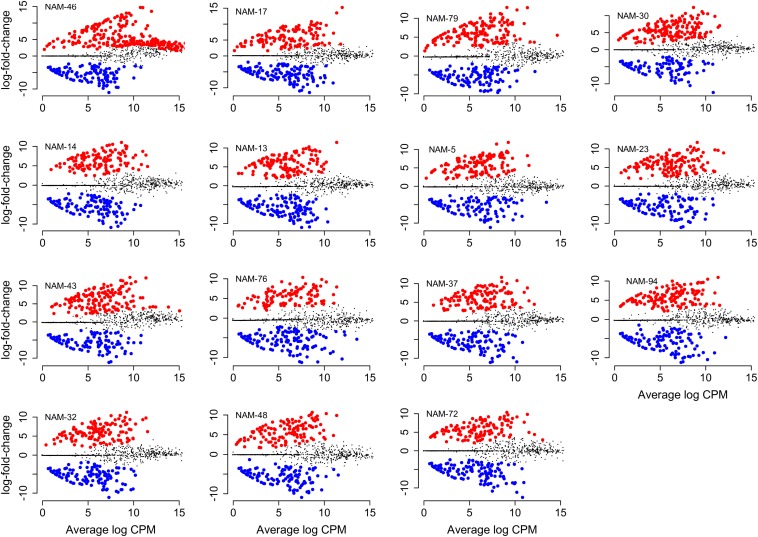
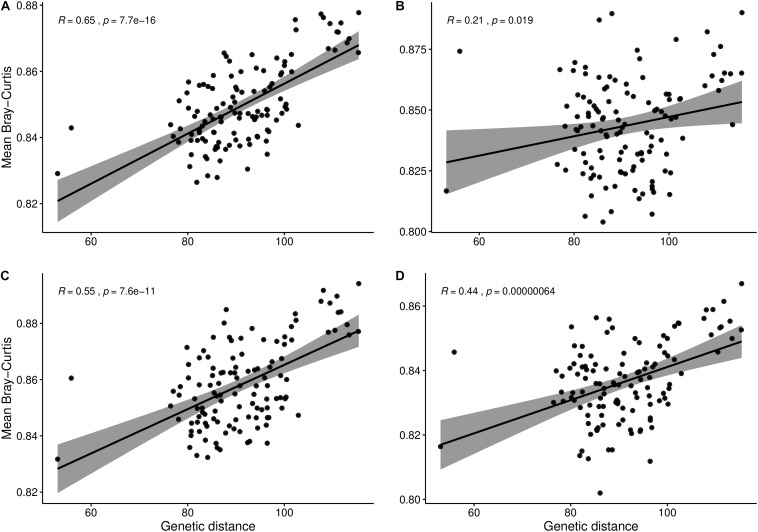
Modifying the rhizosphere microbiome through targeted plant breeding is key to harnessing positive plant-microbial interrelationships in cropping agroecosystems. Here, we examine the composition of rhizosphere bacterial communities of diverse genotypes to identify: (1) taxa that preferentially associate with genotypes, (2) core bacterial microbiota associated with , (3) heritable alpha diversity measures at flowering and whole growing season, and (4) correlation between microbial and plant genetic distance among canola genotypes at different growth stages. Our aim is to identify and describe signature microbiota with potential positive benefits that could be integrated in breeding and management strategies. Rhizosphere soils of 16 diverse genotypes sampled weekly over a 10-week period at single location as well as at three time points at two additional locations were analyzed using 16S rRNA gene amplicon sequencing. The rhizosphere microbiome was characterized by diverse bacterial communities with 32 named bacterial phyla. The most abundant phyla were Proteobacteria, Actinobacteria, and Acidobacteria. Overall microbial and plant genetic distances were highly correlated ( = 0.65). Alpha diversity heritability estimates were between 0.16 and 0.41 when evaluated across growth stage and between 0.24 and 0.59 at flowering. Compared with a reference genotype, a total of 81 genera were significantly more abundant and 71 were significantly less abundant in at least one genotype out of the total 558 bacterial genera. Most differentially abundant genera were Proteobacteria and Actinobacteria followed by Bacteroidetes and Firmicutes. Here, we also show that genotypes select an overall core bacterial microbiome with growth-stage-related patterns as to how taxa joined the core membership. In addition, we report that sets of core taxa were consistent across our three sites and 2 years. Both differential abundance and core analysis implicate numerous bacteria that have been reported to have beneficial effects on plant growth including disease suppression, antifungal properties, and plant growth promotion. Using a multi-site year, temporally intensive field sampling approach, we showed that small plant genetic differences cause predictable changes in canola microbiome and are potential target for direct and indirect selection within breeding programs.
通过定向植物育种来改变根际微生物组是在种植农业生态系统中利用积极的植物 - 微生物相互关系的关键。在此,我们研究了不同基因型根际细菌群落的组成,以确定:(1)优先与基因型相关联的分类群,(2)与之相关的核心细菌微生物群,(3)开花期和整个生长季节的可遗传α多样性指标,以及(4)不同生长阶段油菜基因型之间微生物与植物遗传距离的相关性。我们的目标是识别和描述具有潜在积极益处的标志性微生物群,这些微生物群可整合到育种和管理策略中。在一个地点对16种不同基因型的根际土壤进行了为期10周的每周采样,并在另外两个地点的三个时间点进行采样,使用16S rRNA基因扩增子测序进行分析。根际微生物组的特征是具有32个已命名细菌门的多样细菌群落。最丰富的门是变形菌门、放线菌门和酸杆菌门。总体微生物和植物遗传距离高度相关(= 0.65)。在整个生长阶段评估时,α多样性遗传力估计值在0.16至0.41之间,在开花期为0.24至0.59之间。与一个参考基因型相比,在总共558个细菌属中,共有81个属在至少一种基因型中显著更丰富,71个属显著更不丰富。差异丰度最高的属大多是变形菌门和放线菌门,其次是拟杆菌门和厚壁菌门。在此,我们还表明,基因型选择了一个总体核心细菌微生物组,其分类群加入核心成员的方式具有与生长阶段相关的模式。此外,我们报告说,核心分类群集在我们的三个地点和两年中是一致的。差异丰度分析和核心分析都涉及到许多据报道对植物生长有有益影响的细菌,包括病害抑制、抗真菌特性和植物生长促进。使用多地点多年、时间密集的田间采样方法,我们表明,小的植物遗传差异会导致油菜微生物组的可预测变化,并且是育种计划中直接和间接选择的潜在目标。