Mou Tian, Deng Wenjiang, Gu Fengyun, Pawitan Yudi, Vu Trung Nghia
Department of Medical Epidemiology and Biostatistics, Karolinska Institutet, Stockholm, Sweden.
School of Mathematical Sciences, University College Cork, Cork, Ireland.
Front Genet. 2020 Jan 17;10:1331. doi: 10.3389/fgene.2019.01331. eCollection 2019.
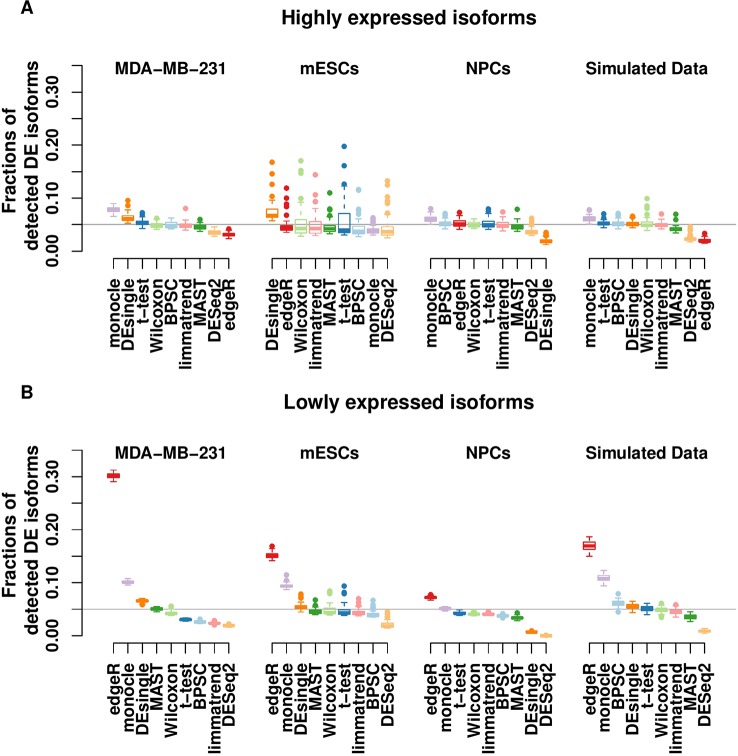
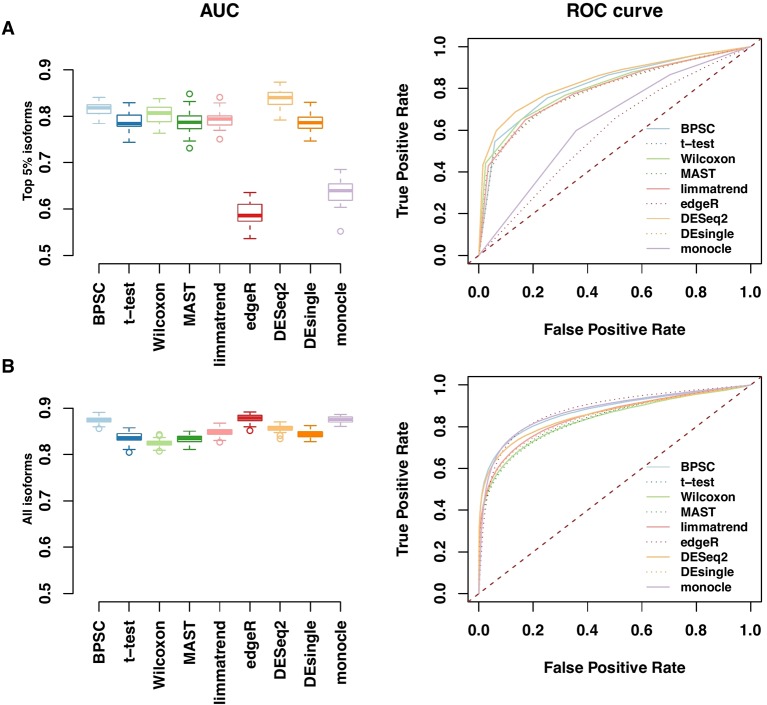
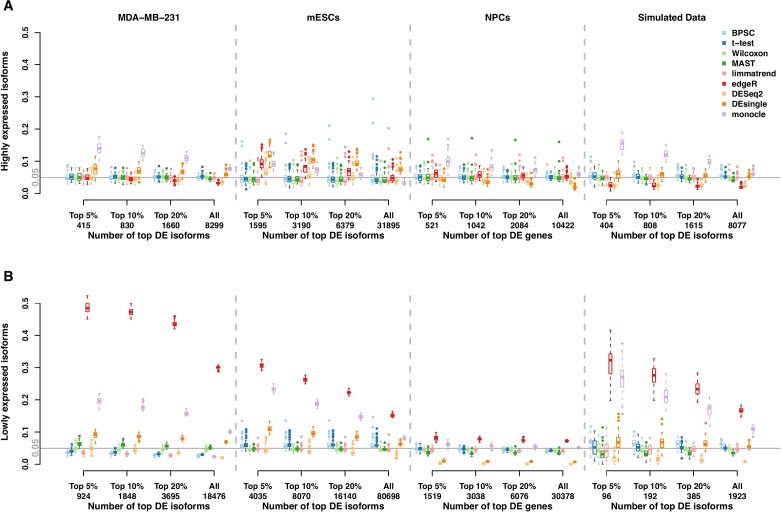
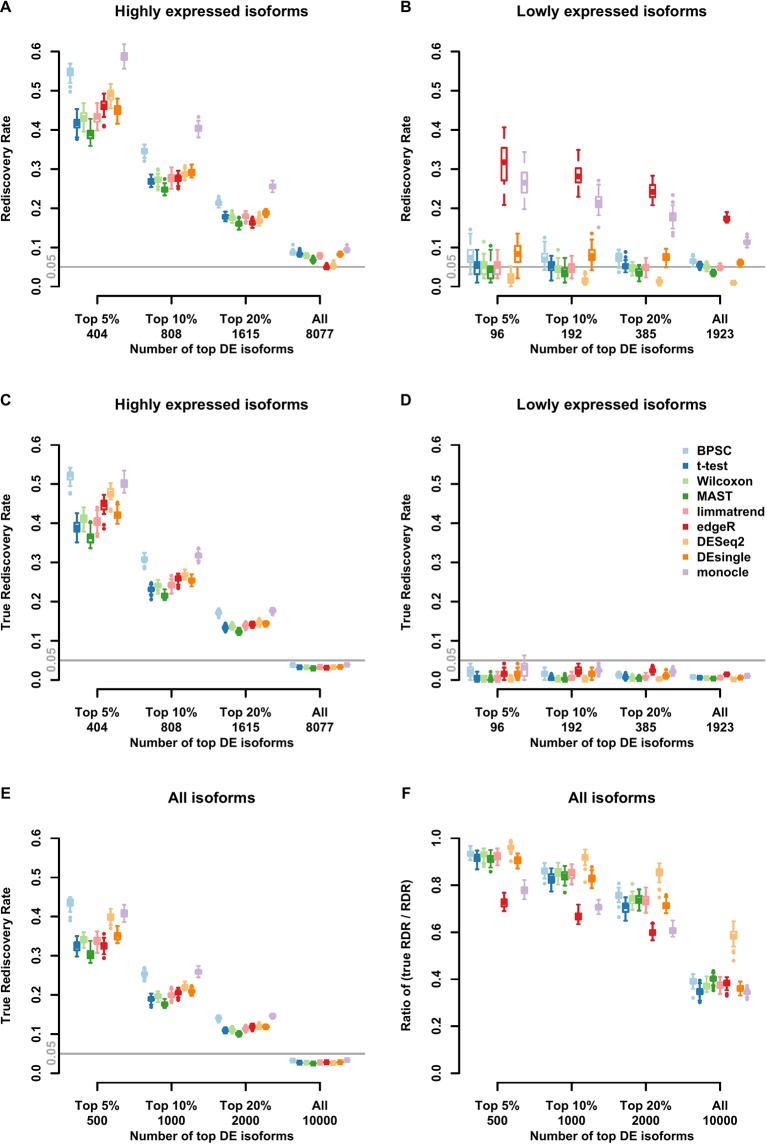
Detection of differentially expressed genes is a common task in single-cell RNA-seq (scRNA-seq) studies. Various methods based on both bulk-cell and single-cell approaches are in current use. Due to the unique distributional characteristics of single-cell data, it is important to compare these methods with rigorous statistical assessments. In this study, we assess the reproducibility of 9 tools for differential expression analysis in scRNA-seq data. These tools include four methods originally designed for scRNA-seq data, three popular methods originally developed for bulk-cell RNA-seq data but have been applied in scRNA-seq analysis, and two general statistical tests. Instead of comparing the performance across all genes, we compare the methods in terms of the rediscovery rates (RDRs) of top-ranked genes, separately for highly and lowly expressed genes. Three real and one simulated scRNA-seq data sets are used for the comparisons. The results indicate that some widely used methods, such as edgeR and monocle, have worse RDR performances compared to the other methods, especially for the top-ranked genes. For highly expressed genes, many bulk-cell-based methods can perform similarly to the methods designed for scRNA-seq data. But for the lowly expressed genes performance varies substantially; edgeR and monocle are too liberal and have poor control of false positives, while DESeq2 is too conservative and consequently loses sensitivity compared to the other methods. BPSC, Limma, DEsingle, MAST, t-test and Wilcoxon have similar performances in the real data sets. Overall, the scRNA-seq based method BPSC performs well against the other methods, particularly when there is a sufficient number of cells.
检测差异表达基因是单细胞RNA测序(scRNA-seq)研究中的一项常见任务。目前使用的各种方法基于批量细胞和单细胞方法。由于单细胞数据独特的分布特征,通过严格的统计评估来比较这些方法很重要。在本研究中,我们评估了9种用于scRNA-seq数据差异表达分析工具的可重复性。这些工具包括四种最初为scRNA-seq数据设计的方法、三种最初为批量细胞RNA-seq数据开发但已应用于scRNA-seq分析的常用方法,以及两种通用统计检验。我们不是比较所有基因的性能,而是分别针对高表达和低表达基因,根据排名靠前基因的重新发现率(RDR)来比较这些方法。使用三个真实和一个模拟的scRNA-seq数据集进行比较。结果表明,一些广泛使用的方法,如edgeR和monocle,与其他方法相比,RDR性能较差,尤其是对于排名靠前的基因。对于高表达基因,许多基于批量细胞的方法与为scRNA-seq数据设计的方法表现相似。但对于低表达基因,性能差异很大;edgeR和monocle过于宽松,对假阳性的控制较差,而DESeq2过于保守,因此与其他方法相比失去了敏感性。BPSC、Limma、DEsingle、MAST、t检验和Wilcoxon在真实数据集中表现相似。总体而言,基于scRNA-seq的方法BPSC与其他方法相比表现良好,特别是在有足够数量细胞的情况下。