Institute for Computational Biomedicine, Bioquant, Heidelberg University, Faculty of Medicine, and Heidelberg University Hospital, Heidelberg, Germany.
Joint Research Centre for Computational Biomedicine (JRC-COMBINE), RWTH Aachen University, Faculty of Medicine, Aachen, Germany.
Genome Biol. 2020 Feb 12;21(1):36. doi: 10.1186/s13059-020-1949-z.
Many functional analysis tools have been developed to extract functional and mechanistic insight from bulk transcriptome data. With the advent of single-cell RNA sequencing (scRNA-seq), it is in principle possible to do such an analysis for single cells. However, scRNA-seq data has characteristics such as drop-out events and low library sizes. It is thus not clear if functional TF and pathway analysis tools established for bulk sequencing can be applied to scRNA-seq in a meaningful way.
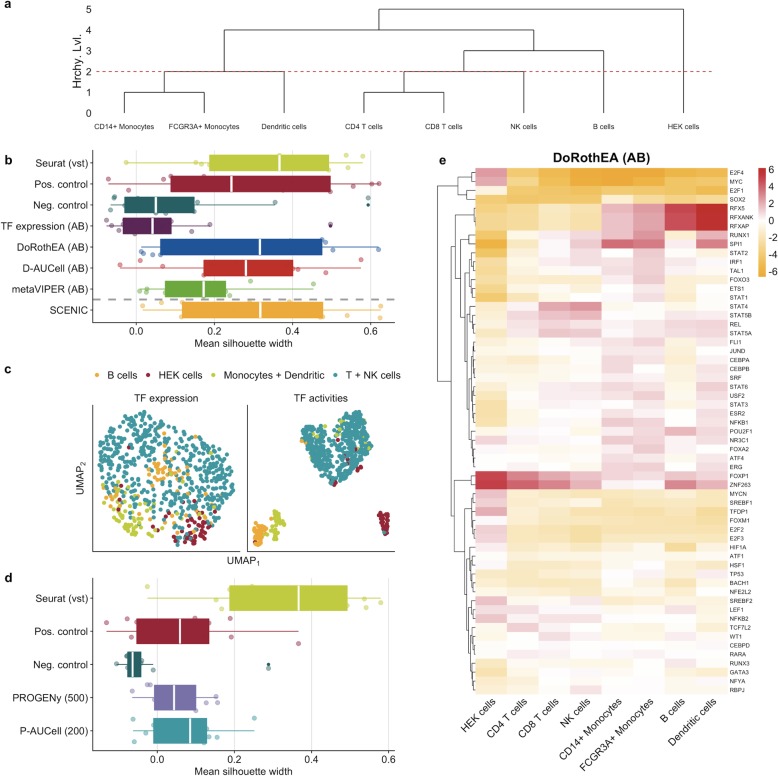
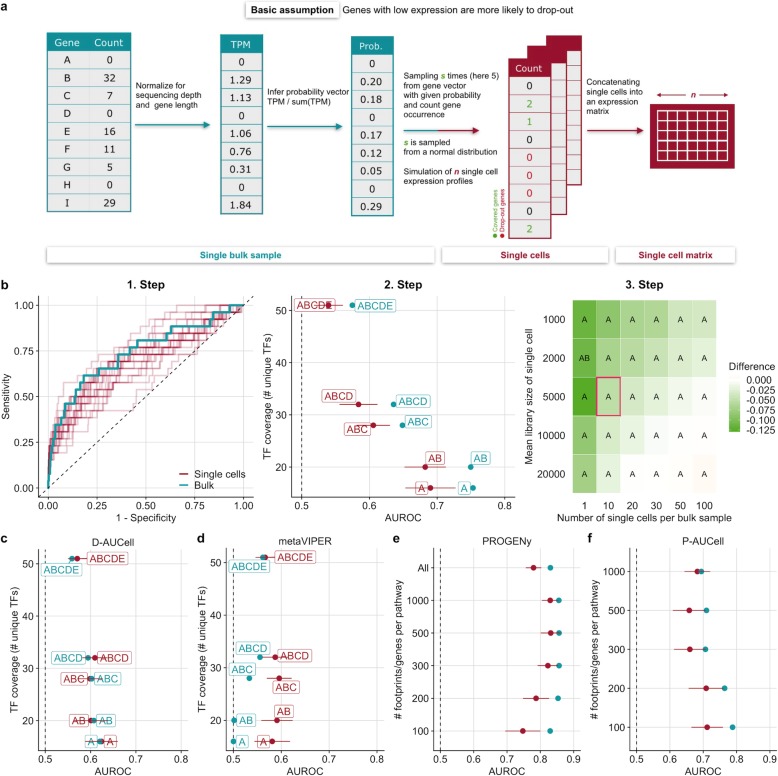
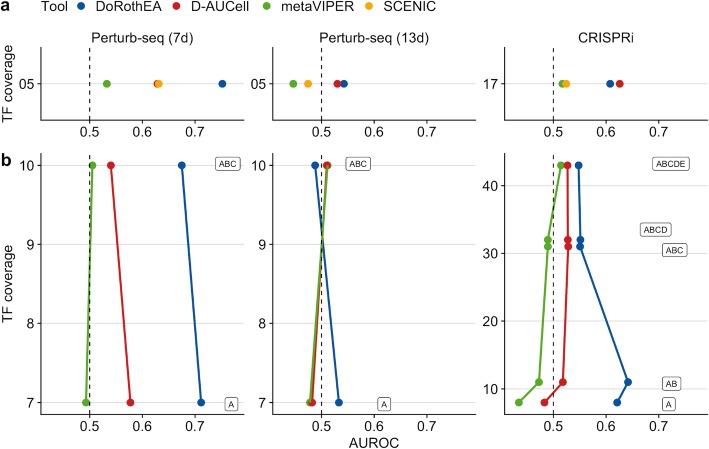
To address this question, we perform benchmark studies on simulated and real scRNA-seq data. We include the bulk-RNA tools PROGENy, GO enrichment, and DoRothEA that estimate pathway and transcription factor (TF) activities, respectively, and compare them against the tools SCENIC/AUCell and metaVIPER, designed for scRNA-seq. For the in silico study, we simulate single cells from TF/pathway perturbation bulk RNA-seq experiments. We complement the simulated data with real scRNA-seq data upon CRISPR-mediated knock-out. Our benchmarks on simulated and real data reveal comparable performance to the original bulk data. Additionally, we show that the TF and pathway activities preserve cell type-specific variability by analyzing a mixture sample sequenced with 13 scRNA-seq protocols. We also provide the benchmark data for further use by the community.
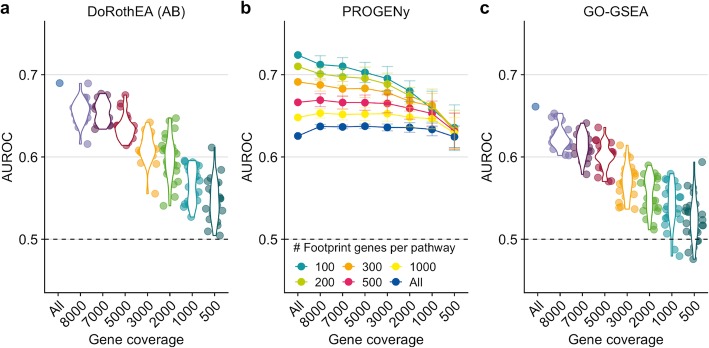
Our analyses suggest that bulk-based functional analysis tools that use manually curated footprint gene sets can be applied to scRNA-seq data, partially outperforming dedicated single-cell tools. Furthermore, we find that the performance of functional analysis tools is more sensitive to the gene sets than to the statistic used.
许多功能分析工具已经被开发出来,以从批量转录组数据中提取功能和机制见解。随着单细胞 RNA 测序(scRNA-seq)的出现,原则上可以对单细胞进行这样的分析。然而,scRNA-seq 数据具有诸如缺失事件和低文库大小等特征。因此,尚不清楚为批量测序建立的功能 TF 和途径分析工具是否可以以有意义的方式应用于 scRNA-seq。
为了解决这个问题,我们在模拟和真实的 scRNA-seq 数据上进行基准研究。我们包括用于估计途径和转录因子(TF)活性的批量 RNA 工具 PROGENy、GO 富集和 DoRothEA,以及专为 scRNA-seq 设计的工具 SCENIC/AUCell 和 metaVIPER,并将它们进行比较。对于计算机研究,我们从 TF/途径扰动批量 RNA-seq 实验中模拟单细胞。我们用 CRISPR 介导的敲除后的真实 scRNA-seq 数据补充模拟数据。我们在模拟和真实数据上的基准测试结果与原始批量数据的性能相当。此外,我们通过分析用 13 种 scRNA-seq 方案测序的混合样本,表明 TF 和途径活性保留了细胞类型特异性的可变性。我们还提供基准数据供社区进一步使用。
我们的分析表明,基于批量的功能分析工具,使用手动编辑的足迹基因集,可以应用于 scRNA-seq 数据,在某些情况下优于专门的单细胞工具。此外,我们发现功能分析工具的性能对基因集比对统计数据更为敏感。