Wu I-Wen, Lin Chan-Yu, Chang Lun-Ching, Lee Chin-Chan, Chiu Chih-Yung, Hsu Heng-Jung, Sun Chiao-Yin, Chen Yuen-Chan, Kuo Yu-Lun, Yang Chi-Wei, Gao Sheng-Siang, Hsieh Wen-Ping, Chung Wen-Hung, Lai Hsin-Chih, Su Shih-Chi
Department of Nephrology, Chang Gung Memorial Hospital, Keelung, Taiwan.
College of Medicine, Chang Gung University, Taoyuan, Taiwan.
Int J Biol Sci. 2020 Jan 1;16(3):420-434. doi: 10.7150/ijbs.37421. eCollection 2020.
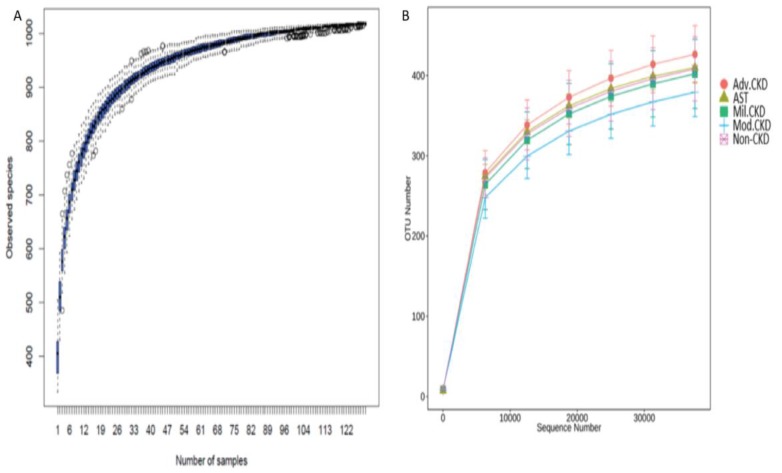
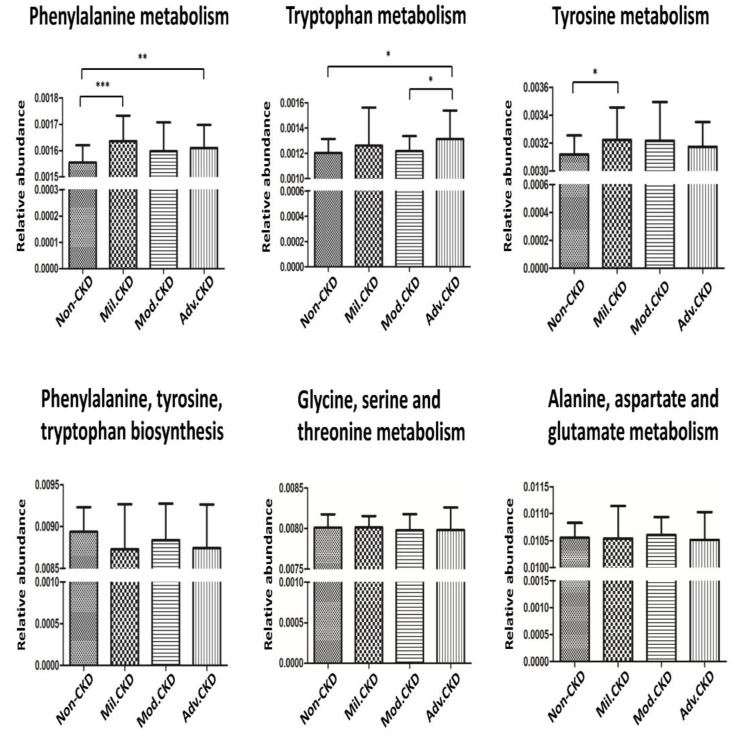
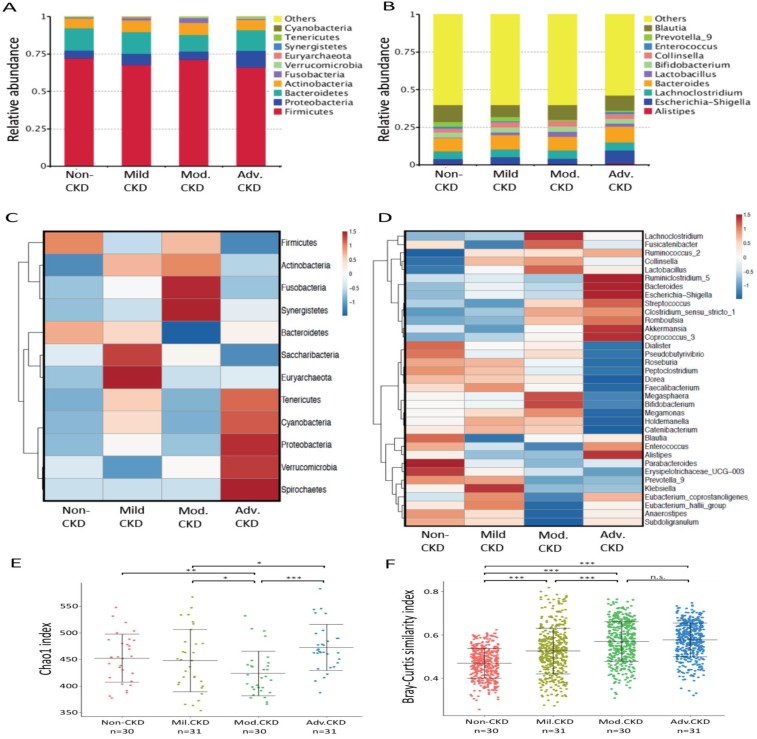
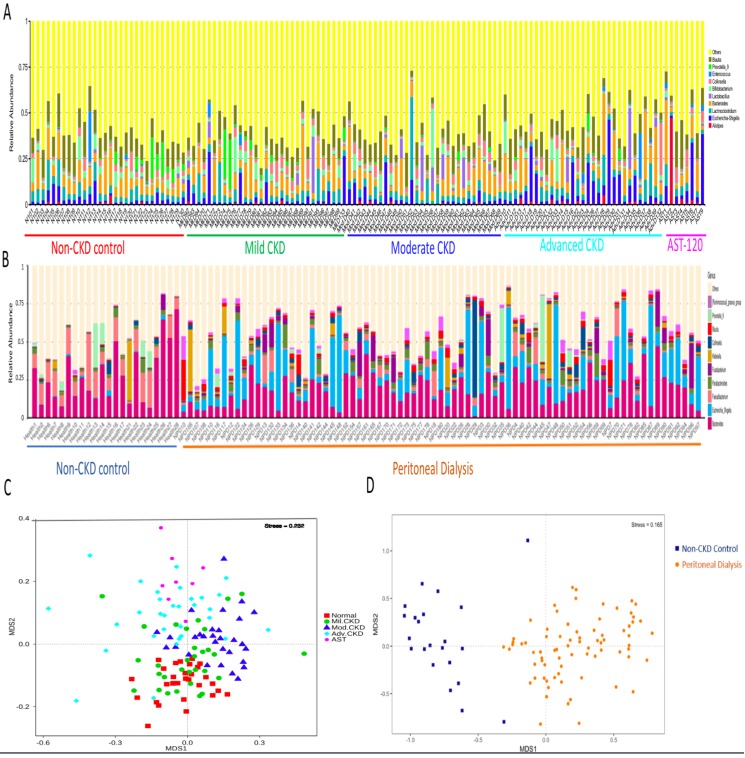
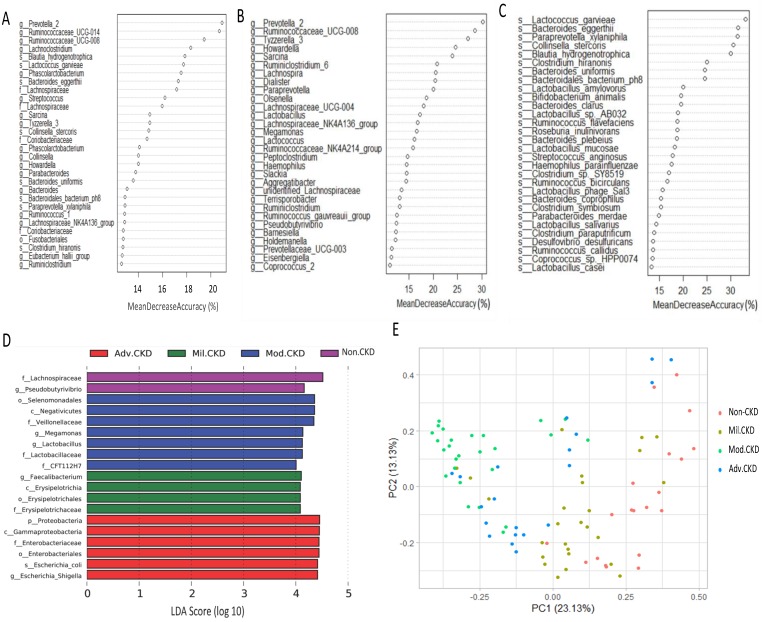
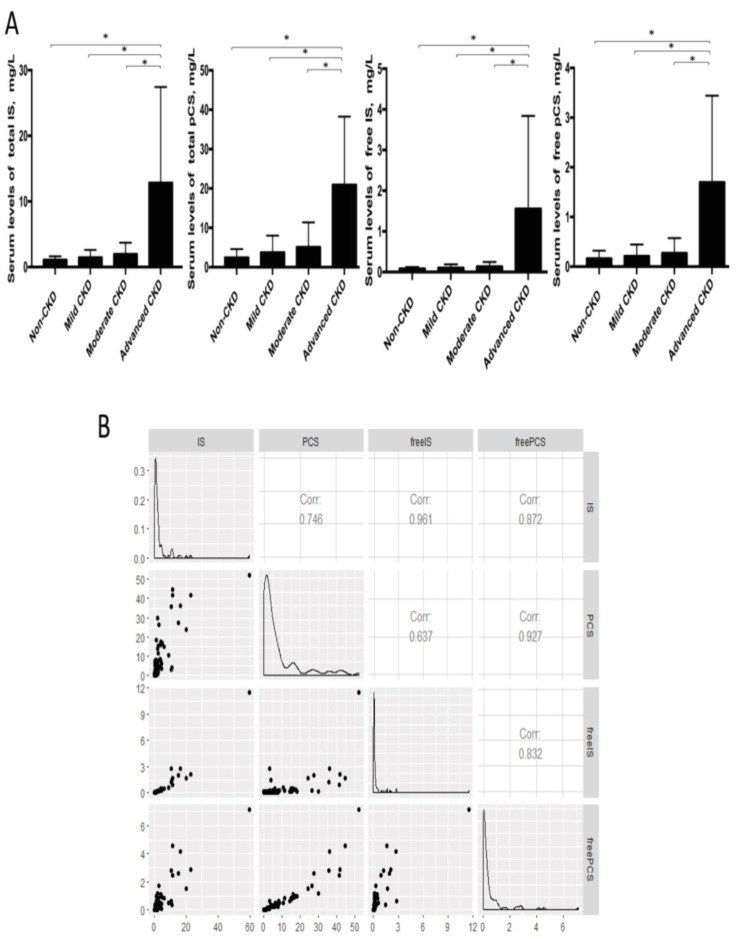
The interplay of the gut microbes with gut-producing nephrotoxins and the renal progression remains unclear in large human cohort. Significant compositional and functional differences in the intestinal microbiota (by 16S rRNA gene sequencing) were noted among 30 controls and 92 (31 mild, 30 moderate and 31 advanced) patients at different chronic kidney disease (CKD) stages (discovery cohort). A core CKD-associated microbiota consisted of 7 genera (Escherichia_Shigella, Dialister, Lachnospiraceae_ND3007_group, Pseudobutyrivibrio, Roseburia, Paraprevotella and Ruminiclostridium) and 2 species (Collinsella stercoris and Bacteroides eggerthii) were identified to be highly correlated with the stages of CKD. Paraprevotella, Pseudobutyrivibrio and Collinsella stercoris were superior in discriminating CKD from the controls than the use of urine protein/creatinine ratio, even at early-stage of disease. The performance was further confirmed in a validation cohort comprising 22 controls and 76 peritoneal dialysis patients. Bacterial genera highly correlated with indoxyl sulfate and p-cresyl sulfate levels were identified. Prediction of the functional capabilities of microbial communities showed that microbial genes related to the metabolism of aromatic amino acids (phenylalanine, tyrosine, and tryptophan) were differentially enriched among the control and different CKD stages. Collectively, our results provide solid human evidence of the impact of gut-metabolite-kidney axis on the severity of chronic kidney disease and highlight a usefulness of specific gut microorganisms as possible disease differentiate marker of this global health burden.
在大型人类队列中,肠道微生物与肠道产生的肾毒素之间的相互作用以及肾脏进展情况仍不清楚。在30名对照者和92名处于不同慢性肾脏病(CKD)阶段(发现队列)的患者(31名轻度、30名中度和31名重度)中,通过16S rRNA基因测序发现肠道微生物群存在显著的组成和功能差异。一个与CKD相关的核心微生物群由7个属(埃希氏菌属-志贺氏菌属、戴阿李斯特菌属、毛螺菌科ND3007组、假丁酸弧菌属、罗斯氏菌属、副普雷沃氏菌属和瘤胃梭菌属)和2个种(粪便柯林斯菌和埃氏拟杆菌)组成,它们被确定与CKD阶段高度相关。与使用尿蛋白/肌酐比值相比,副普雷沃氏菌属、假丁酸弧菌属和粪便柯林斯菌在区分CKD与对照者方面表现更优,即使在疾病早期也是如此。在一个由22名对照者和76名腹膜透析患者组成的验证队列中,这一表现得到了进一步证实。确定了与硫酸吲哚酚和对甲酚硫酸盐水平高度相关的细菌属。对微生物群落功能能力的预测表明,与芳香族氨基酸(苯丙氨酸、酪氨酸和色氨酸)代谢相关的微生物基因在对照者和不同CKD阶段之间存在差异富集。总体而言,我们的结果为肠道代谢物-肾脏轴对慢性肾脏病严重程度的影响提供了确凿的人体证据,并突出了特定肠道微生物作为这种全球健康负担可能的疾病鉴别标志物的有用性。