Abramyan Ara M, Yano Hideaki, Xu Min, Liu Leanne, Naing Sett, Fant Andrew D, Shi Lei
Computational Chemistry and Molecular Biophysics Unit, National Institute on Drug Abuse - Intramural Research Program, National Institutes of Health, Baltimore, MD, United States.
Comput Struct Biotechnol J. 2020 Jan 7;18:199-206. doi: 10.1016/j.csbj.2019.12.012. eCollection 2020.
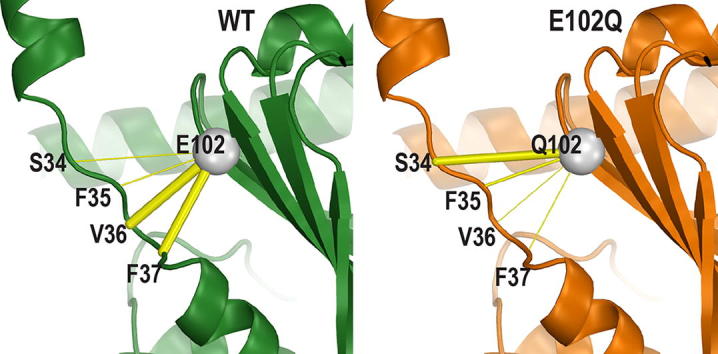
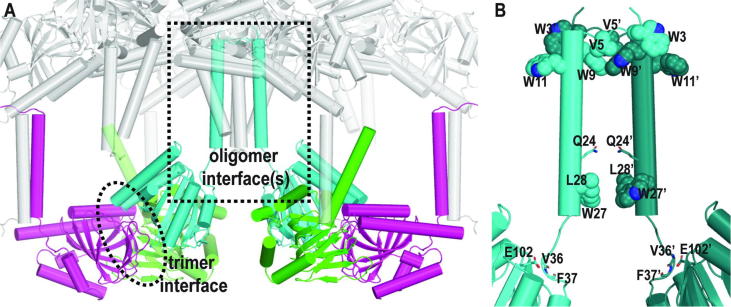
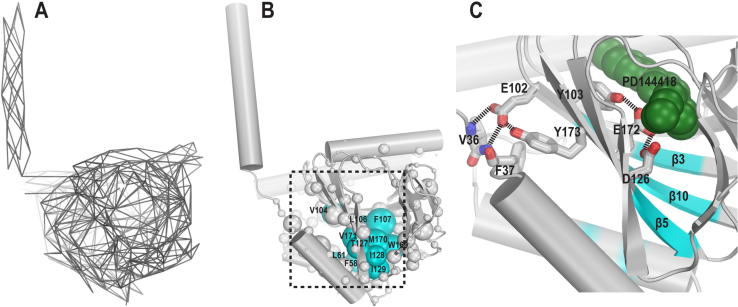
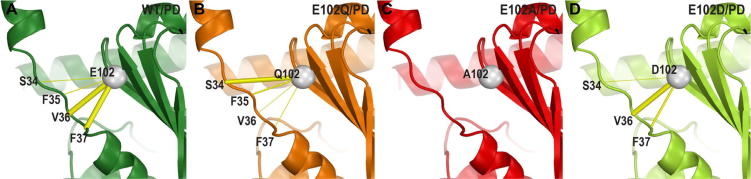
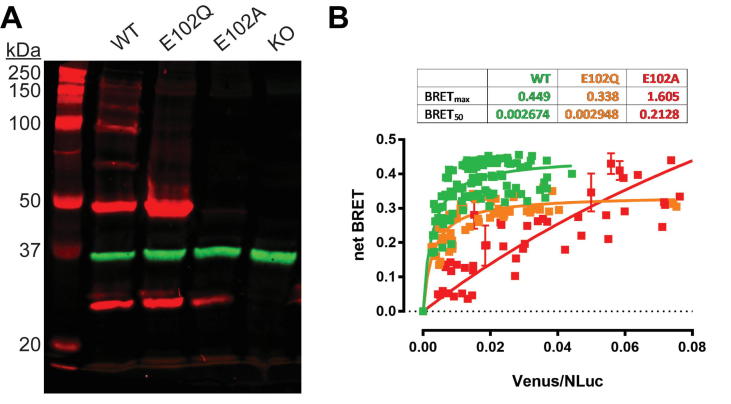
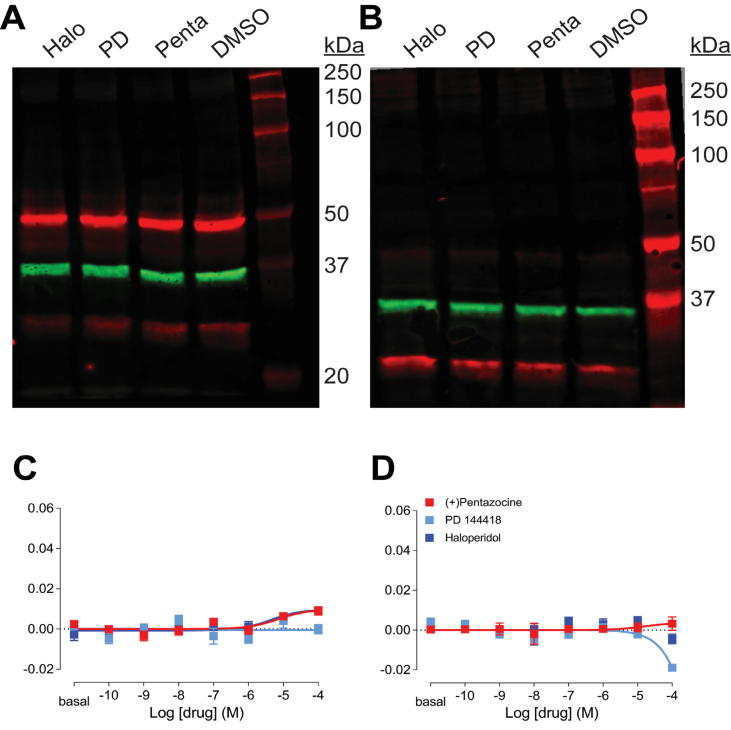
The sigma 1 receptor (σ1R) is a unique endoplasmic reticulum membrane protein. Its ligands have been shown to possess therapeutic potential for neurological and substance use disorders among others. The E102Q mutation of σ1R has been found to elicit familial cases of amyotrophic lateral sclerosis (ALS). Despite reports of its downstream signaling consequences, the mechanistic details of the functional impact of E102Q at molecular level are not clear. Here, we investigate the molecular mechanism of the E102Q mutation with a spectrum of biochemical, biophysical, and pharmacological approaches. Our analysis of the interaction network of σ1R indicates that a set of residues near E102 is critical for the integrity of C-terminal ligand-binding domain. However, this integrity is not affected by the E102Q and E102A mutations, which is confirmed by the radioligand binding results. Instead, the E102 mutations disrupt the connection between the C-terminal domain and the N-terminal transmembrane helix (NT-helix). Results from bioluminescence resonance energy transfer and western blot assays demonstrate that these mutations destabilize higher-order σ1R oligomers, while our molecular dynamics simulations based on a σ1R crystal structure reveal a potential mechanism by which the mutations perturb the NT-helix dynamics. Thus, we propose that E102 is at a critical position in propagating the effects of ligand binding from the C-terminal domain to the NT-helix, while the latter may be involved in forming alternative oligomer interfaces, separate from the previously reported trimer interface. Together, these results provide the first account of the molecular mechanism of σ1R dysfunction caused by E102Q.
σ1受体(σ1R)是一种独特的内质网膜蛋白。其配体已被证明对神经疾病和物质使用障碍等具有治疗潜力。已发现σ1R的E102Q突变会引发家族性肌萎缩侧索硬化症(ALS)病例。尽管有关于其下游信号传导后果的报道,但E102Q在分子水平上功能影响的机制细节尚不清楚。在这里,我们用一系列生物化学、生物物理和药理学方法研究E102Q突变的分子机制。我们对σ1R相互作用网络的分析表明,E102附近的一组残基对C端配体结合域的完整性至关重要。然而,这种完整性不受E102Q和E102A突变的影响,放射性配体结合结果证实了这一点。相反,E102突变破坏了C端结构域与N端跨膜螺旋(NT-螺旋)之间的连接。生物发光共振能量转移和蛋白质印迹分析结果表明,这些突变使高阶σ1R寡聚体不稳定,而我们基于σ1R晶体结构的分子动力学模拟揭示了这些突变扰乱NT-螺旋动力学的潜在机制。因此,我们提出E102在将配体结合的效应从C端结构域传递到NT-螺旋中处于关键位置,而后者可能参与形成与先前报道的三聚体界面不同的替代寡聚体界面。总之,这些结果首次阐述了由E102Q引起的σ1R功能障碍的分子机制。