Sato Shuji, Uemoto Yoshinobu, Kikuchi Takashi, Egawa Sachiko, Kohira Kimiko, Saito Tomomi, Sakuma Hironori, Miyashita Satoshi, Arata Shinji, Kojima Takatoshi, Suzuki Keiichi
National Livestock Breeding Center, Nishigo, Fukushima, 961-8511, Japan.
Miyazaki Branch of National Livestock Breeding Center, Kobayashi, Miyazaki, 886-0004, Japan.
BMC Genet. 2016 Apr 19;17:60. doi: 10.1186/s12863-016-0368-3.
The aim of the present study was to compare the power of single nucleotide polymorphism (SNP)-based genome-wide association study (GWAS) and haplotype-based GWAS for quantitative trait loci (QTL) detection, and to detect novel candidate genes affecting economically important traits in a purebred Duroc population comprising seven-generation pedigree. First, we performed a simulation analysis using real genotype data of this population to compare the power (based on the null hypothesis) of the two methods. We then performed GWAS using both methods and real phenotype data comprising 52 traits, which included growth, carcass, and meat quality traits.
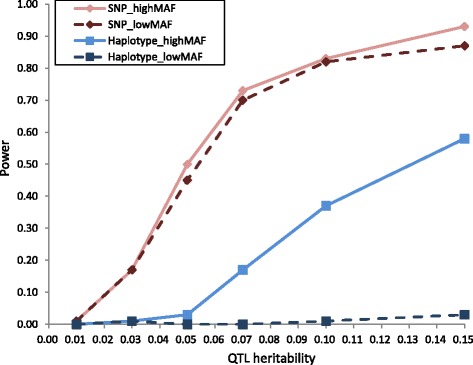
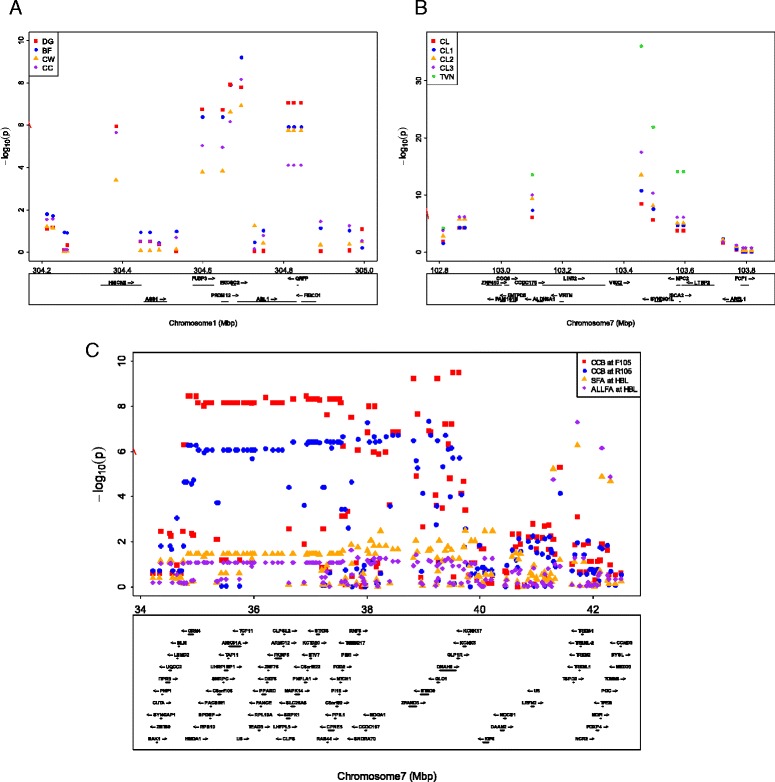
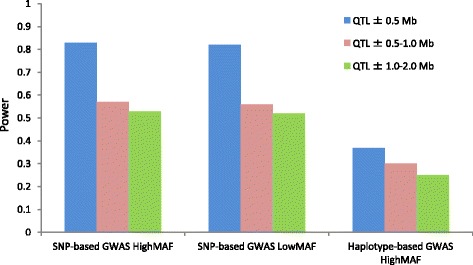
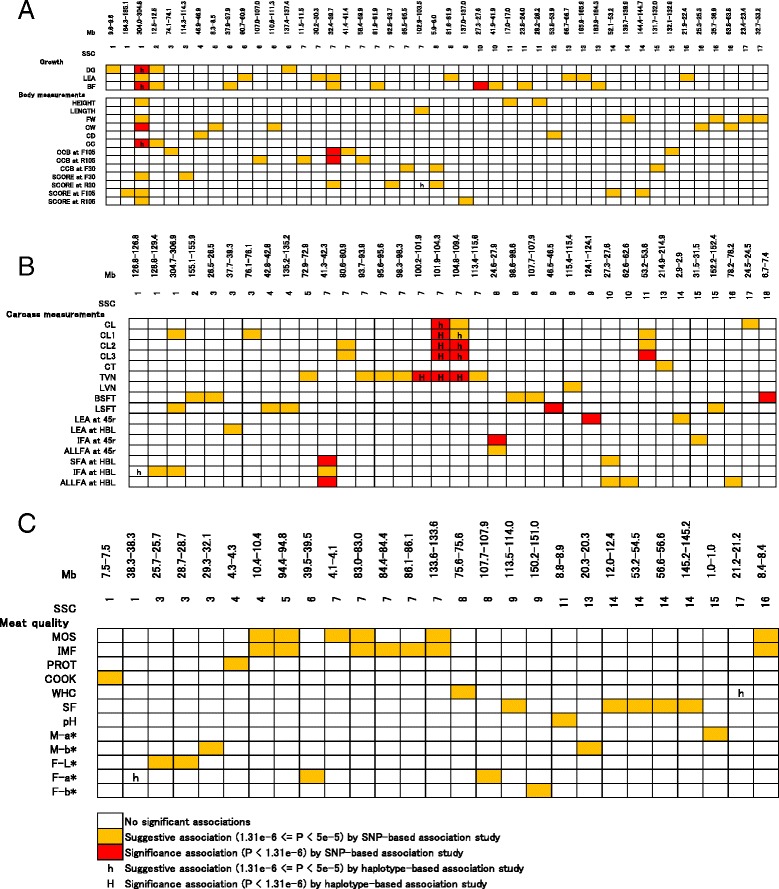
In total, 836 animals were genotyped using the Illumina PorcineSNP60 BeadChip and 14 customized SNPs from regions of known candidate genes related to the traits of interest. The power of SNP-based GWAS was greater than that of haplotype-based GWAS in a simulation analysis. In real data analysis, a larger number of significant regions was obtained by SNP-based GWAS than by haplotype-based GWAS. For SNP-based GWAS, 23 genome-wide significant SNP regions were detected for 17 traits, and 120 genome-wide suggestive SNP regions were detected for 27 traits. For haplotype-based GWAS, 6 genome-wide significant SNP regions were detected for four traits, and 11 genome-wide suggestive SNP regions were detected for eight traits. All genome-wide significant SNP regions detected by haplotype-based GWAS were located in regions also detected by SNP-based GWAS. Four regions detected by SNP-based GWAS were significantly associated with multiple traits: on Sus scrofa chromosome (SSC) 1 at 304 Mb; and on SSC7 at 35-39 Mb, 41-42 Mb, and 103 Mb. The vertnin gene (VRTN) in particular, was located on SSC7 at 103 Mb and was significantly associated with vertebrae number and carcass lengths. Mapped QTL regions contain some candidate genes involved in skeletal formation (FUBP3; far upstream element binding protein 3) and fat deposition (METTL3; methyltransferase like 3).
Our results show that a multigenerational pig population is useful for detecting QTL, which are typically segregated in a purebred population. In addition, a novel significant region could be detected by SNP-based GWAS as opposed to haplotype-based GWAS.
本研究的目的是比较单核苷酸多态性(SNP)为基础的全基因组关联研究(GWAS)和单倍型为基础的GWAS在检测数量性状基因座(QTL)方面的效能,并在一个包含七代系谱的纯种杜洛克猪群体中检测影响经济重要性状的新候选基因。首先,我们使用该群体的真实基因型数据进行模拟分析,以比较两种方法的效能(基于零假设)。然后,我们使用这两种方法以及包含52个性状的真实表型数据进行GWAS分析,这些性状包括生长、胴体和肉质性状。
总共使用Illumina PorcineSNP60 BeadChip和来自与感兴趣性状相关的已知候选基因区域的14个定制SNP对836头动物进行了基因分型。在模拟分析中,基于SNP的GWAS的效能大于基于单倍型的GWAS。在实际数据分析中,基于SNP的GWAS比基于单倍型的GWAS获得了更多的显著区域。对于基于SNP的GWAS,针对17个性状检测到23个全基因组显著的SNP区域,针对27个性状检测到120个全基因组提示性SNP区域。对于基于单倍型的GWAS,针对4个性状检测到6个全基因组显著的SNP区域,针对8个性状检测到11个全基因组提示性SNP区域。基于单倍型的GWAS检测到的所有全基因组显著SNP区域都位于基于SNP的GWAS也检测到的区域。基于SNP的GWAS检测到的4个区域与多个性状显著相关:在猪1号染色体(SSC1)上304 Mb处;以及在SSC7上35 - 39 Mb、41 - 42 Mb和103 Mb处。特别是,vertnin基因(VRTN)位于SSC7上103 Mb处,与椎骨数量和胴体长度显著相关。定位的QTL区域包含一些参与骨骼形成(FUBP3;远上游元件结合蛋白3)和脂肪沉积(METTL3;甲基转移酶样3)的候选基因。
我们的结果表明,一个多代猪群体对于检测QTL是有用的,这些QTL通常在纯种群体中分离。此外,与基于单倍型的GWAS相比,基于SNP的GWAS可以检测到一个新的显著区域。