Department of Nephrology and Medical Intensive Care, Charité, Universitätsmedizin Berlin, Berlin, Germany.
Medizinische Klinik and Poliklinik IV, Klinikum der Universität München, Munich, Germany.
J Biomed Sci. 2020 Feb 14;27(1):34. doi: 10.1186/s12929-020-0623-9.
In the past years evidence has been growing about the interconnection of chronic kidney disease and acute kidney injury. The underlying pathophysiological mechanisms remain unclear. We hypothesized, that a threshold ischemia time in unilateral ischemia/reperfusion injury sets an extent of ischemic tubule necrosis, which as "point of no return" leads to progressive injury. This progress is temporarily associated by increased markers of inflammation and results in fibrosis and atrophy of the ischemic kidney.
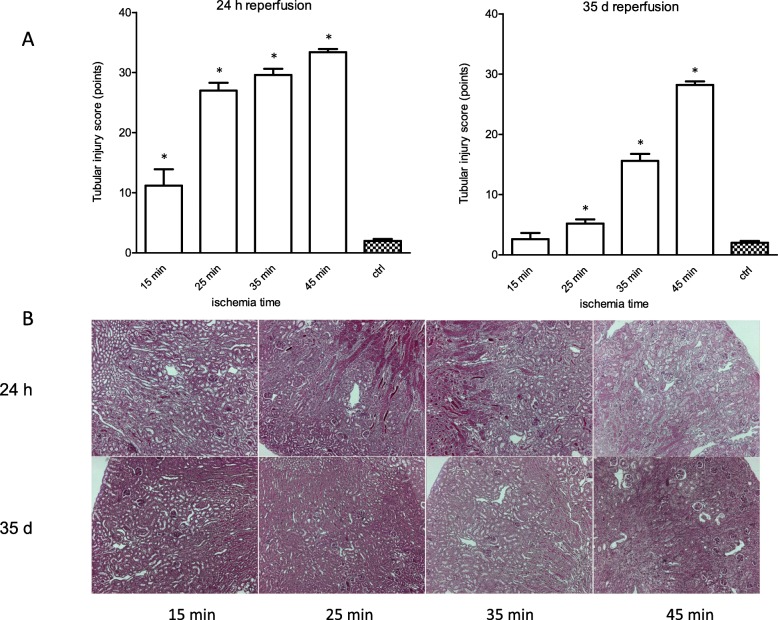
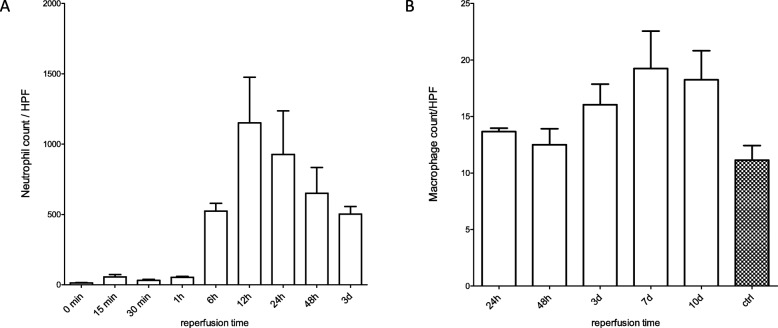
Acute tubule necrosis was induced by unilateral ischemia/reperfusion injury in male C57BL/6 N mice with different ischemia times (15, 25, 35, and 45 min). At multiple time points between 15 min and 5 weeks we assessed gene expression of markers for injury, inflammation, and fibrosis, histologically the injury of tubules, cell death (TUNEL), macrophages, neutrophil influx and kidney atrophy.
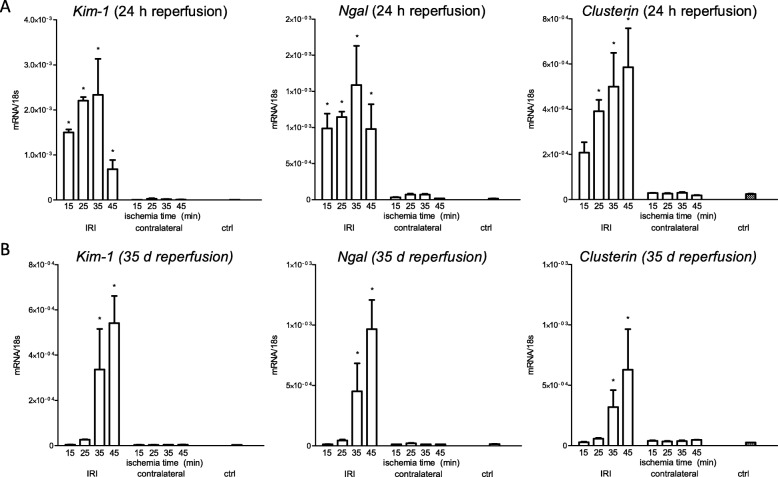
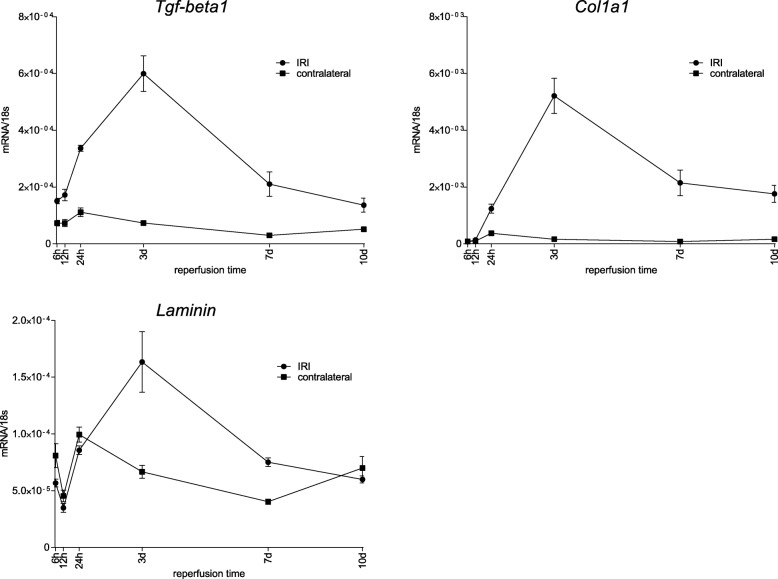
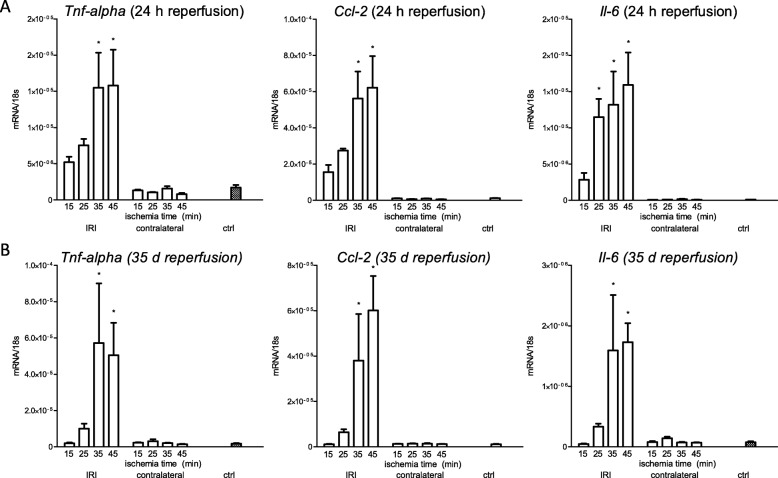
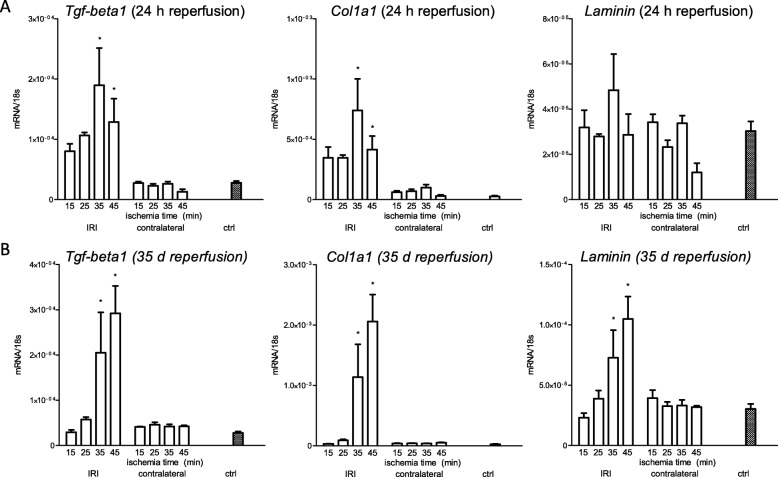
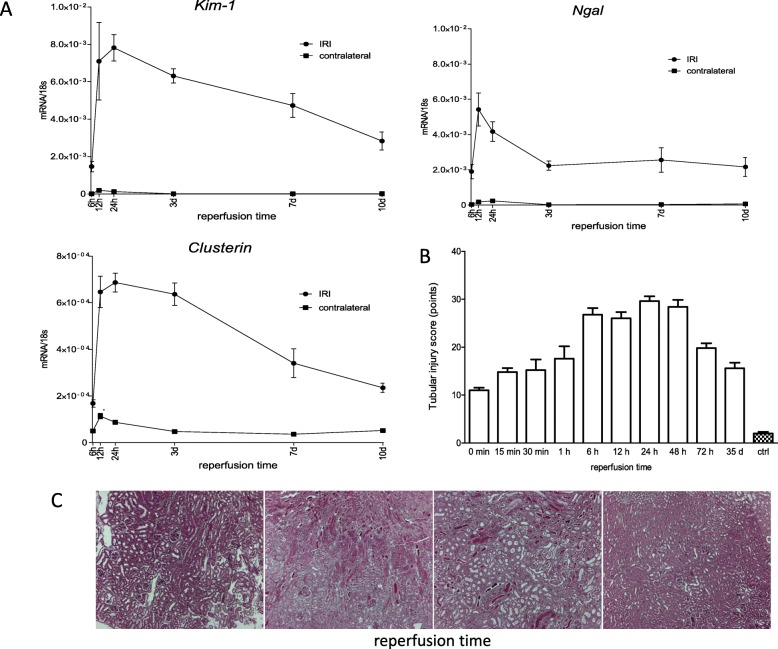
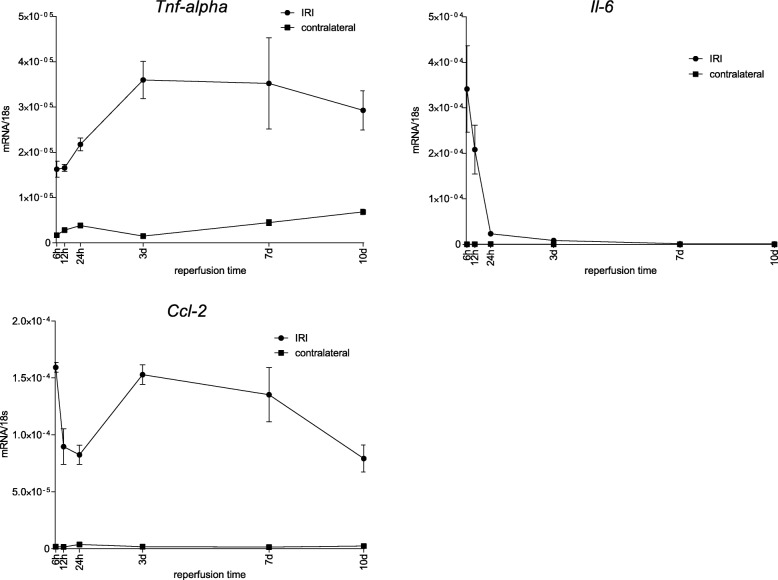
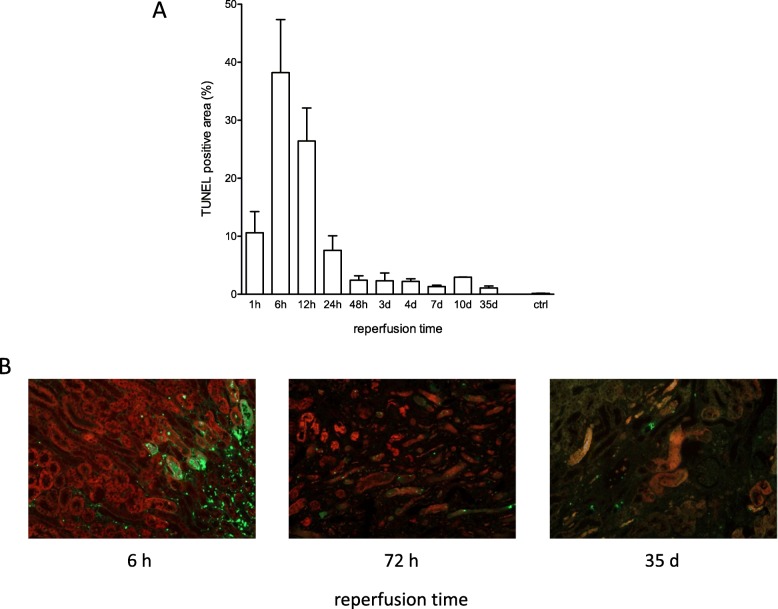
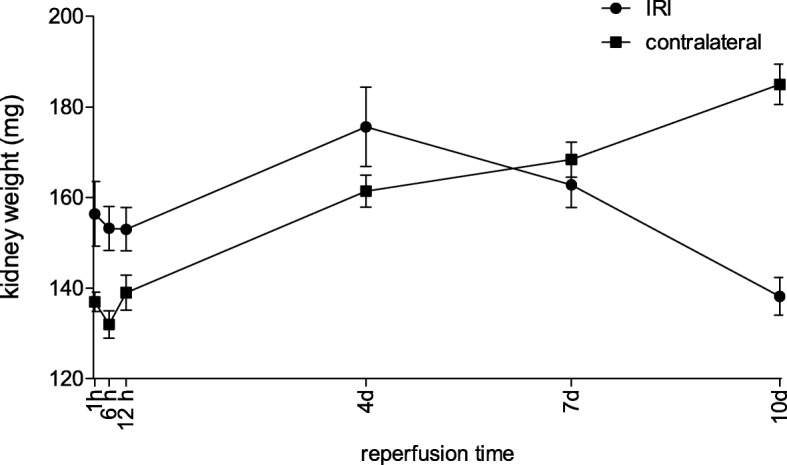
Unilateral ischemia for 15 and 25 min induced upregulation of markers for injury after reperfusion for 24 h but no upregulation after 5 weeks. None of the markers for inflammation or fibrosis were upregulated after ischemia for 15 and 25 min at 24 h or 5 weeks on a gene expression level, except for Il-6. Ischemia for 35 and 45 min consistently induced upregulation of markers for inflammation, injury, and partially of fibrosis (Tgf-β1 and Col1a1) at 24 h and 5 weeks. The threshold ischemia time for persistent injury of 35 min induced a temporal association of markers for inflammation and injury with peaks between 6 h and 7 d along the course of 10 d. This ischemia time also induced persistent cell death (TUNEL) throughout observation for 5 weeks with a peak at 6 h and progressing kidney atrophy beginning 7 d after ischemia.
This study confirms the evidence of a threshold extent of ischemic injury in which markers of injury, inflammation and fibrosis do not decline to baseline but remain upregulated assessed in long term outcome (5 weeks). Excess of this threshold as "point of no return" leads to persistent cell death and progressing atrophy and is characterized by a temporal association of markers for inflammation and injury.
近年来,有证据表明慢性肾脏病和急性肾损伤之间存在关联。其潜在的病理生理机制尚不清楚。我们假设,单侧缺血/再灌注损伤中的缺血时间阈值会导致一定程度的缺血性肾小管坏死,这一“不可逆转点”会导致进行性损伤。这种进展暂时与炎症标志物的增加有关,并导致缺血肾脏的纤维化和萎缩。
通过单侧缺血/再灌注损伤在雄性 C57BL/6N 小鼠中诱导急性肾小管坏死,缺血时间分别为 15、25、35 和 45 分钟。在 15 分钟至 5 周之间的多个时间点,我们评估了损伤、炎症和纤维化标志物的基因表达,组织学上评估了肾小管损伤、细胞死亡(TUNEL)、巨噬细胞、中性粒细胞浸润和肾脏萎缩。
15 分钟和 25 分钟的单侧缺血在再灌注 24 小时后诱导了损伤标志物的上调,但在 5 周后没有上调。在基因表达水平上,除了 Il-6 之外,15 分钟和 25 分钟的缺血在 24 小时和 5 周时没有引起炎症或纤维化标志物的上调。35 分钟和 45 分钟的缺血始终在 24 小时和 5 周时诱导炎症、损伤和部分纤维化标志物(Tgf-β1 和 Col1a1)的上调。35 分钟的持续缺血损伤的时间阈值导致炎症和损伤标志物在 10 天的过程中在 6 小时至 7 天之间出现高峰,并伴有持续的细胞死亡(TUNEL)。这种缺血时间还导致持续的细胞死亡(TUNEL)在 5 周的观察期间,在 6 小时时达到峰值,并在缺血后 7 天开始进行性肾萎缩。
本研究证实了缺血损伤程度存在阈值的证据,在长期结果(5 周)中,损伤、炎症和纤维化标志物不会降至基线水平,而是持续上调。超过这个阈值的“不可逆转点”会导致持续的细胞死亡和进行性萎缩,并以炎症和损伤标志物的时间相关性为特征。