INSERM U973, Laboratory MTi, University Paris Diderot, Paris, France.
Laboratory SABNP, University of Evry, INSERM U1204, Université Paris-Saclay, Evry, France.
Mol Genet Genomic Med. 2020 Apr;8(4):e1166. doi: 10.1002/mgg3.1166. Epub 2020 Feb 25.
Different types of in silico approaches can be used to predict the phenotypic consequence of missense variants. Such algorithms are often categorized as sequence based or structure based, when they necessitate 3D structural information. In addition, many other in silico tools, not dedicated to the analysis of variants, can be used to gain additional insights about the possible mechanisms at play.
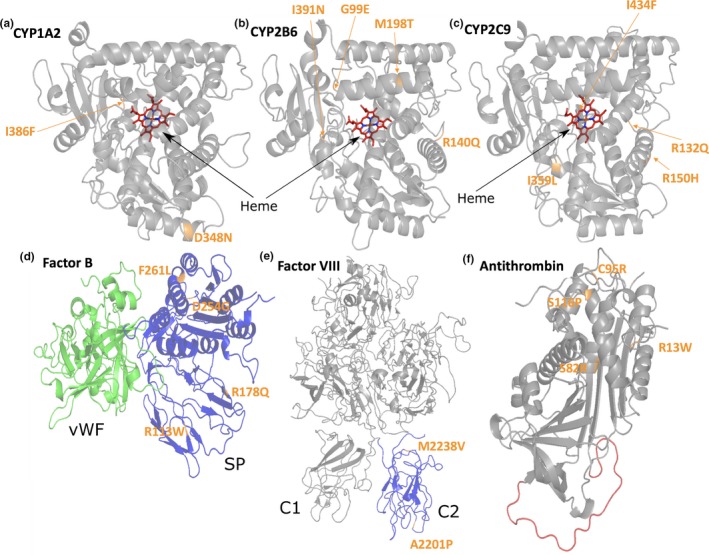
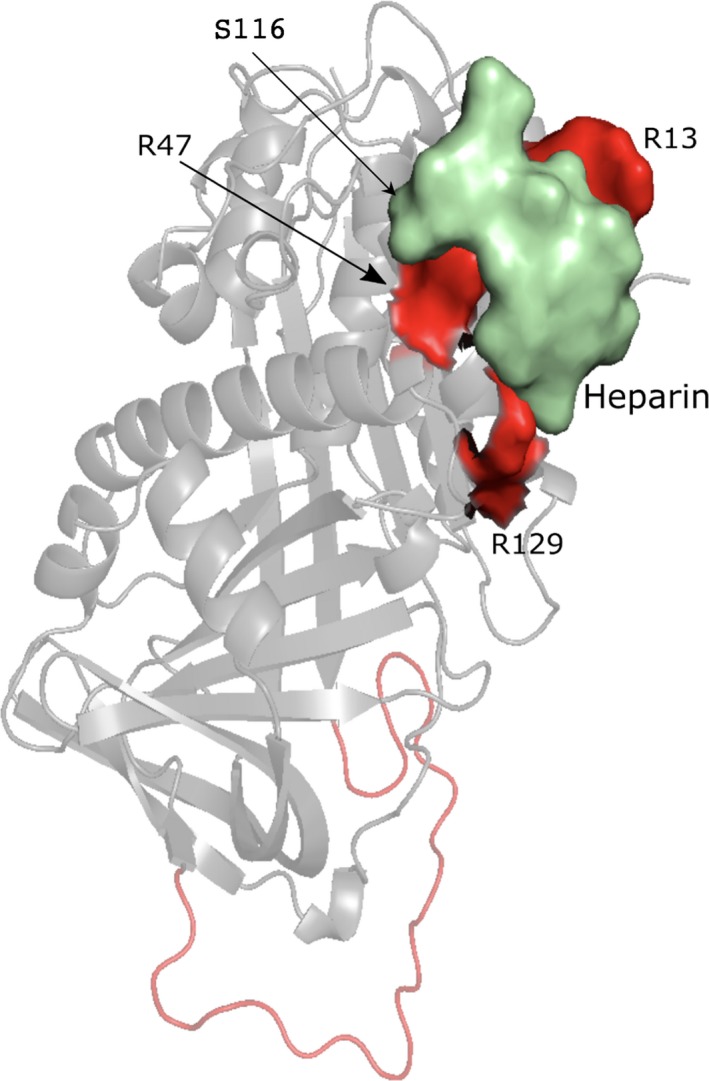
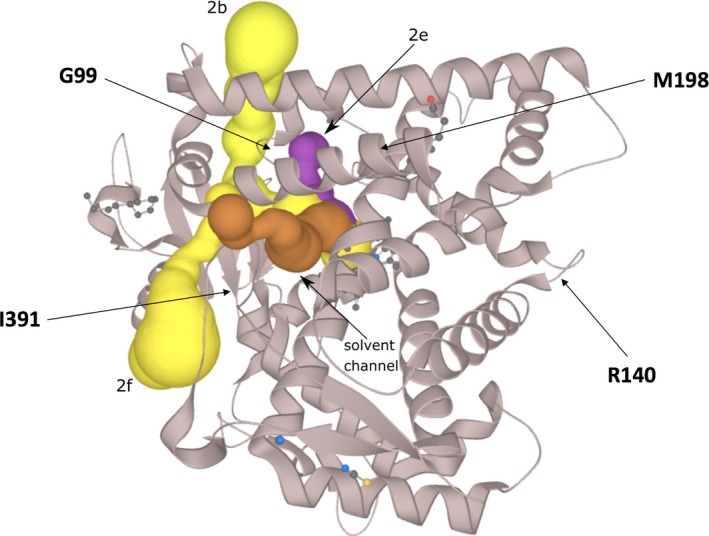
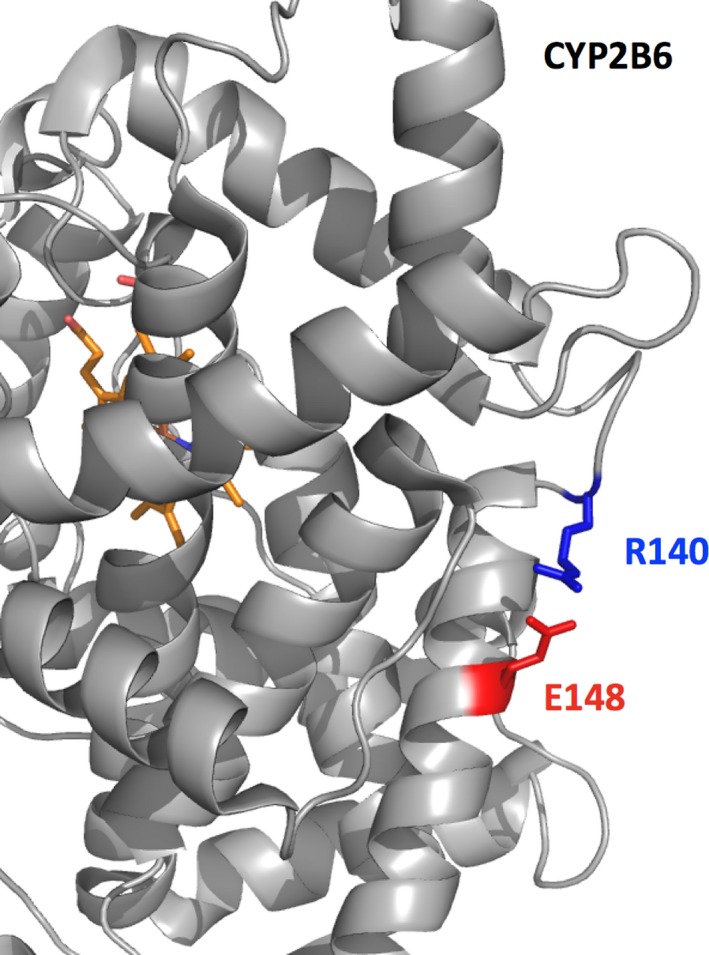
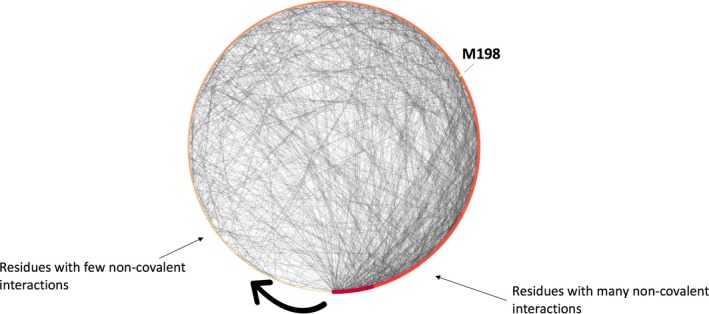
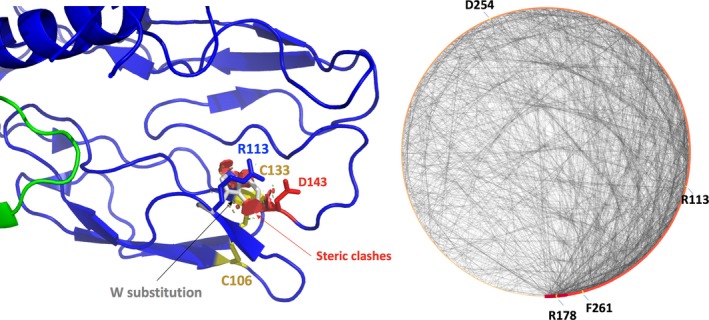
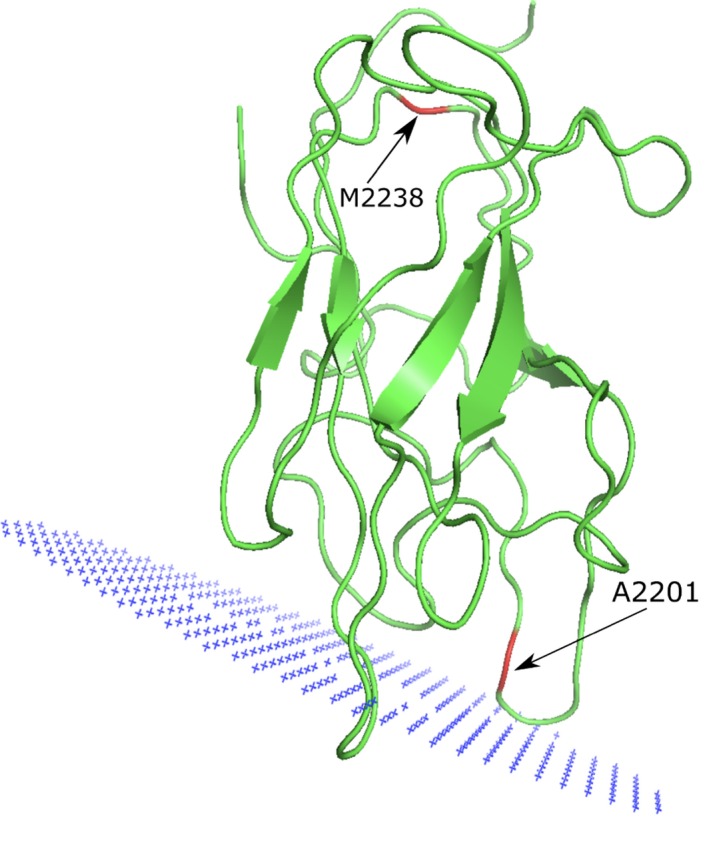
Here we applied different computational approaches to a set of 20 known missense variants present on different proteins (CYP, complement factor B, antithrombin and blood coagulation factor VIII). The tools that were used include fast computational approaches and web servers such as PolyPhen-2, PopMusic, DUET, MaestroWeb, SAAFEC, Missense3D, VarSite, FlexPred, PredyFlexy, Clustal Omega, meta-PPISP, FTMap, ClusPro, pyDock, PPM, RING, Cytoscape, and ChannelsDB.
We observe some conflicting results among the methods but, most of the time, the combination of several engines helped to clarify the potential impacts of the amino acid substitutions.
Combining different computational approaches including some that were not developed to investigate missense variants help to predict the possible impact of the amino acid substitutions. Yet, when the modified residues are involved in a salt-bridge, the tools tend to fail, even when the analysis is performed in 3D. Thus, interactive structural analysis with molecular graphics packages such as Chimera or PyMol or others are still needed to clarify automatic prediction.
不同类型的计算方法可用于预测错义变异的表型后果。这些算法通常分为基于序列或基于结构的,当它们需要 3D 结构信息时。此外,许多其他非专门用于分析变异的计算工具也可用于获得有关可能发挥作用的机制的其他见解。
我们将不同的计算方法应用于一组存在于不同蛋白质(CYP、补体因子 B、抗凝血酶和凝血因子 VIII)上的 20 个已知错义变体。使用的工具包括快速计算方法和 Web 服务器,如 PolyPhen-2、PopMusic、DUET、MaestroWeb、SAAFEC、Missense3D、VarSite、FlexPred、PredyFlexy、Clustal Omega、meta-PPISP、FTMap、ClusPro、pyDock、PPM、RING、Cytoscape 和 ChannelsDB。
我们观察到方法之间存在一些相互矛盾的结果,但大多数情况下,结合使用多个引擎有助于澄清氨基酸取代的潜在影响。
结合使用不同的计算方法,包括一些不是为研究错义变体而开发的方法,有助于预测氨基酸取代的可能影响。然而,当修饰的残基涉及盐桥时,即使在 3D 中进行分析,工具也往往会失败。因此,仍然需要使用 Chimera 或 PyMol 或其他分子图形软件包进行交互式结构分析,以澄清自动预测。