Ottawa Research and Development Centre, Agriculture and Agri-Food Canada, Ottawa, ON K1A 0C6, Canada.
Department of Mathematics and Statistics, University of Waterloo, Waterloo, ON N2L 3G1, Canada.
Int J Mol Sci. 2020 Feb 25;21(5):1577. doi: 10.3390/ijms21051577.
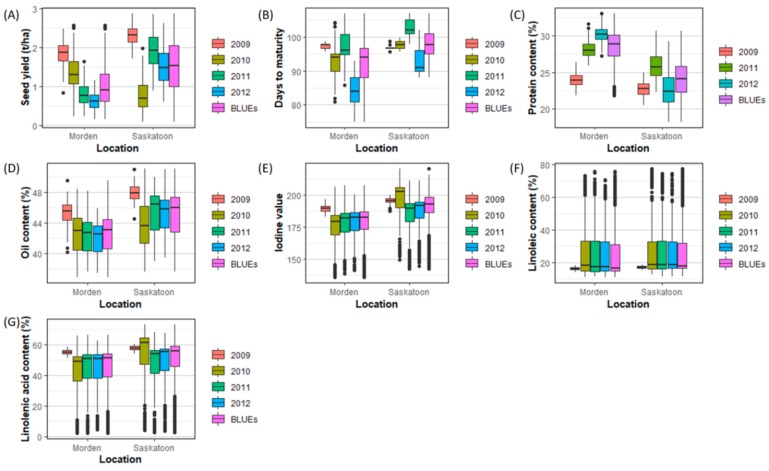
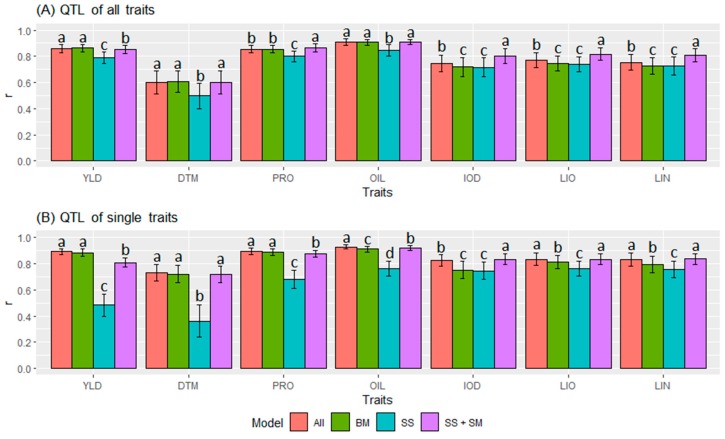
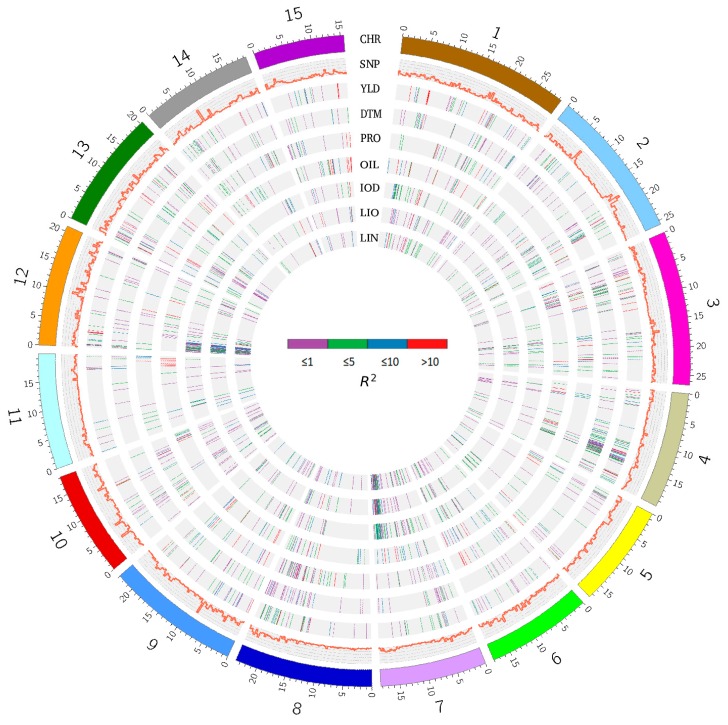
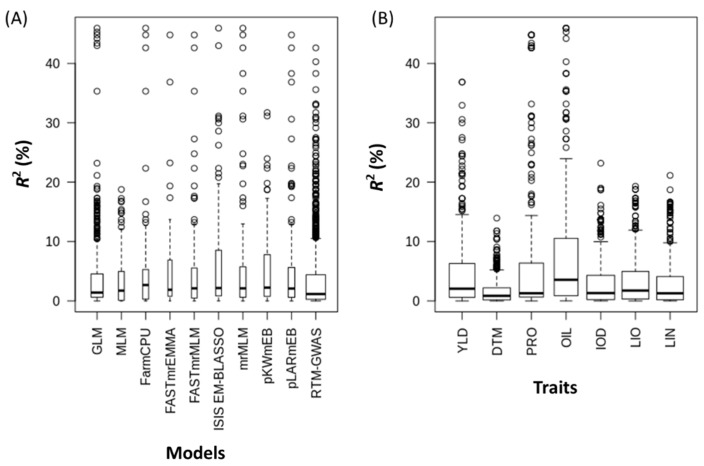
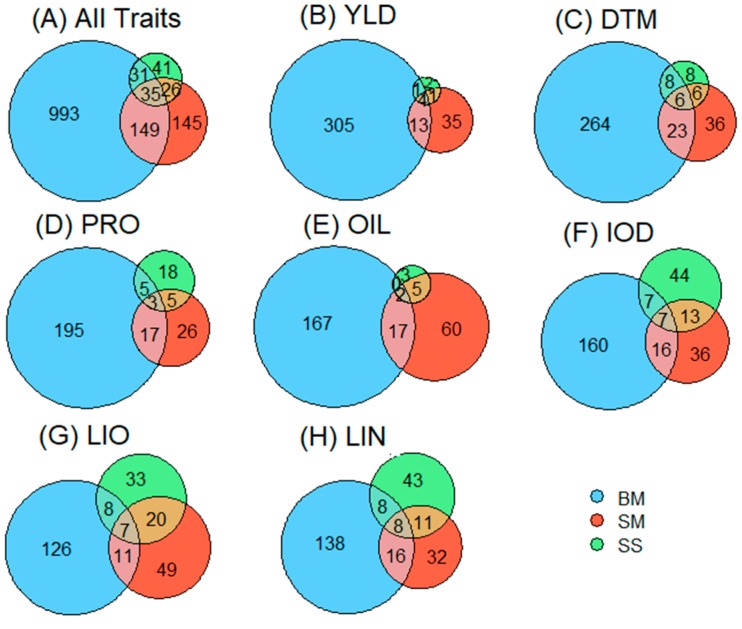
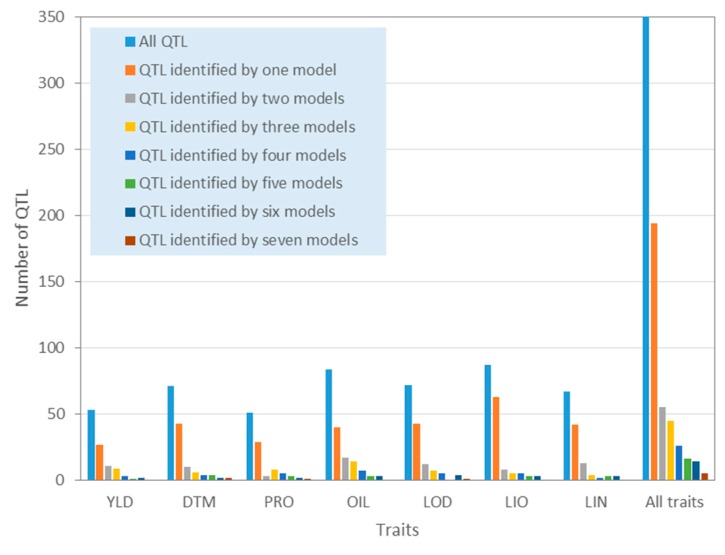
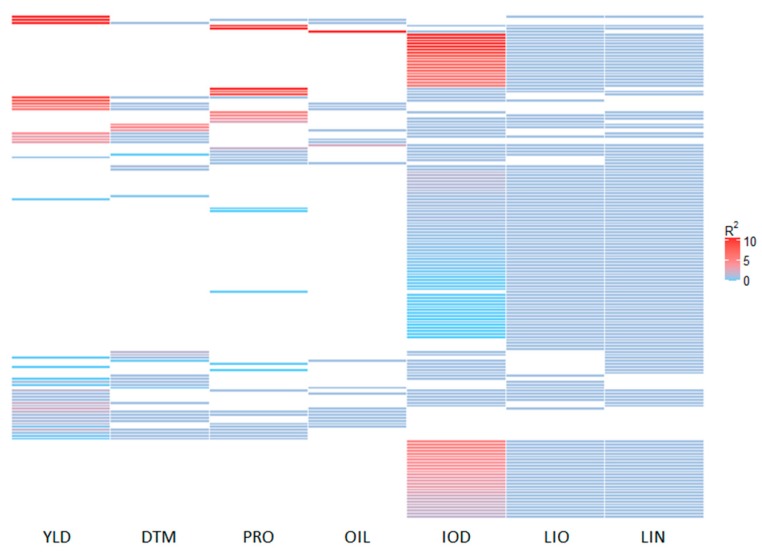
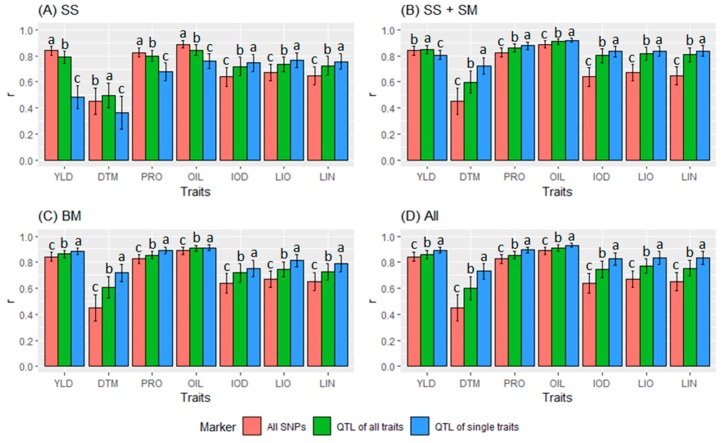
Molecular markers are one of the major factors affecting genomic prediction accuracy and the cost of genomic selection (GS). Previous studies have indicated that the use of quantitative trait loci (QTL) as markers in GS significantly increases prediction accuracy compared with genome-wide random single nucleotide polymorphism (SNP) markers. To optimize the selection of QTL markers in GS, a set of 260 lines from bi-parental populations with 17,277 genome-wide SNPs were used to evaluate the prediction accuracy for seed yield (YLD), days to maturity (DTM), iodine value (IOD), protein (PRO), oil (OIL), linoleic acid (LIO), and linolenic acid (LIN) contents. These seven traits were phenotyped over four years at two locations. Identification of quantitative trait nucleotides (QTNs) for the seven traits was performed using three types of statistical models for genome-wide association study: two SNP-based single-locus (SS), seven SNP-based multi-locus (SM), and one haplotype-block-based multi-locus (BM) models. The identified QTNs were then grouped into QTL based on haplotype blocks. For all seven traits, 133, 355, and 1,208 unique QTL were identified by SS, SM, and BM, respectively. A total of 1420 unique QTL were obtained by SS+SM+BM, ranging from 254 (OIL, LIO) to 361 (YLD) for individual traits, whereas a total of 427 unique QTL were achieved by SS+SM, ranging from 56 (YLD) to 128 (LIO). SS models alone did not identify sufficient QTL for GS. The highest prediction accuracies were obtained using single-trait QTL identified by SS+SM+BM for OIL (0.929 ± 0.016), PRO (0.893 ± 0.023), YLD (0.892 ± 0.030), and DTM (0.730 ± 0.062), and by SS+SM for LIN (0.837 ± 0.053), LIO (0.835 ± 0.049), and IOD (0.835 ± 0.041). In terms of the number of QTL markers and prediction accuracy, SS+SM outperformed other models or combinations thereof. The use of all SNPs or QTL of all seven traits significantly reduced the prediction accuracy of traits. The results further validated that QTL outperformed high-density genome-wide random markers, and demonstrated that the combined use of single and multi-locus models can effectively identify a comprehensive set of QTL that improve prediction accuracy, but further studies on detection and removal of redundant or false-positive QTL to maximize prediction accuracy and minimize the number of QTL markers in GS are warranted.
分子标记物是影响基因组预测准确性和基因组选择(GS)成本的主要因素之一。先前的研究表明,与全基因组随机单核苷酸多态性(SNP)标记物相比,使用数量性状位点(QTL)作为 GS 中的标记物可显著提高预测准确性。为了优化 GS 中 QTL 标记物的选择,使用来自具有 17,277 个全基因组 SNP 的双亲群体的 260 条系评估了种子产量(YLD)、成熟天数(DTM)、碘值(IOD)、蛋白质(PRO)、油(OIL)、亚油酸(LIO)和亚麻酸(LIN)含量的预测准确性。这七个性状在两个地点的四年内进行了表型测定。使用全基因组关联研究的三种统计模型(两种 SNP 为基础的单基因座(SS)、七种 SNP 为基础的多基因座(SM)和一种基于单倍型块的多基因座(BM)模型)对这七个性状的数量性状核苷酸(QTN)进行了鉴定。然后,根据单倍型块将鉴定出的 QTN 分组为 QTL。对于所有七个性状,通过 SS、SM 和 BM 分别鉴定出 133、355 和 1,208 个独特的 QTL。通过 SS+SM+BM 总共获得了 1420 个独特的 QTL,单个性状的范围从 254(OIL,LIO)到 361(YLD),通过 SS+SM 总共获得了 427 个独特的 QTL,范围从 56(YLD)到 128(LIO)。SS 模型本身不足以用于 GS。使用 SS+SM+BM 鉴定的单基因座 QTL 对 OIL(0.929 ± 0.016)、PRO(0.893 ± 0.023)、YLD(0.892 ± 0.030)和 DTM(0.730 ± 0.062)进行了单性状最佳预测,通过 SS+SM 对 LIN(0.837 ± 0.053)、LIO(0.835 ± 0.049)和 IOD(0.835 ± 0.041)进行了单基因座最佳预测。就 QTL 标记物的数量和预测准确性而言,SS+SM 优于其他模型或其组合。使用所有七个性状的所有 SNP 或 QTL 会显著降低性状的预测准确性。结果进一步验证了 QTL 优于高密度全基因组随机标记物,并表明单基因座和多基因座模型的组合使用可以有效地识别出一整套 QTL,从而提高预测准确性,但需要进一步研究检测和去除冗余或假阳性 QTL,以最大程度地提高预测准确性并最小化 GS 中的 QTL 标记物数量。