Zhu Leilei, Li Gongze, Guo Dongliang, Li Xiao, Xue Min, Jiang Haixia, Yan Qingcheng, Xie Fang, Ning Xuefei, Xie Liqiong
Xinjiang Key Laboratory of Biological Resources and Genetic Engineering, College of Life Science and Technology, Xinjiang University, Urumqi, China.
State Key Laboratory of Crop Stress Adaptation and Improvement, School of Life Sciences, Henan University, Zhengzhou, China.
Front Plant Sci. 2024 May 28;15:1403276. doi: 10.3389/fpls.2024.1403276. eCollection 2024.
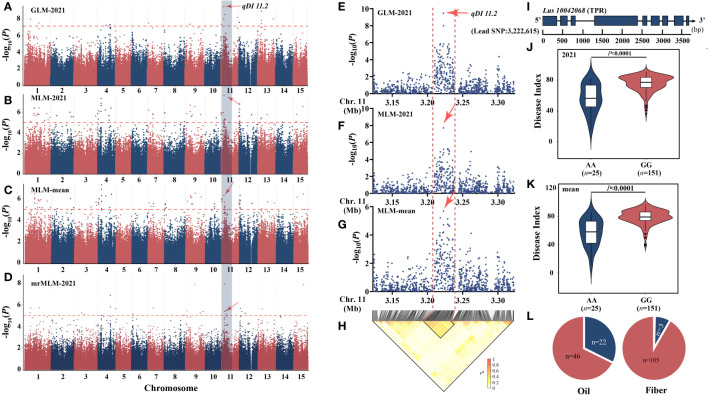
Flax powdery mildew (PM), caused by , is a globally distributed fungal disease of flax, and seriously impairs its yield and quality. To data, only three resistance genes and a few putative quantitative trait loci (QTL) have been reported for flax PM resistance. To dissect the resistance mechanism against PM and identify resistant genetic regions, based on four years of phenotypic datasets (2017, 2019 to 2021), a genome-wide association study (GWAS) was performed on 200 flax core accessions using 674,074 SNPs and 7 models. A total of 434 unique quantitative trait nucleotides (QTNs) associated with 331 QTL were detected. Sixty-four loci shared in at least two datasets were found to be significant in haplotype analyses, and 20 of these sites were shared by multiple models. Simultaneously, a large-effect locus () was detected repeatedly, which was present in the mapping study of flax pasmo resistance loci. Oil flax had more QTL with positive-effect or favorable alleles (PQTL) and showed higher PM resistance than fiber flax, indicating that effects of these QTL were mainly additive. Furthermore, an excellent resistant variety C120 was identified and can be used to promote planting. Based on 331 QTLs identified through GWAS and the statistical model GBLUP, a genomic selection (GS) model related to flax PM resistance was constructed, and the prediction accuracy rate was 0.96. Our results provide valuable insights into the genetic basis of resistance and contribute to the advancement of breeding programs.
亚麻白粉病(PM)由[病原体名称缺失]引起,是一种在全球范围内分布的亚麻真菌病害,严重影响其产量和品质。截至目前,关于亚麻抗白粉病,仅报道了三个抗性基因和一些假定的数量性状位点(QTL)。为剖析亚麻对白粉病的抗性机制并鉴定抗性遗传区域,基于四年(2017年、2019年至2021年)的表型数据集,利用674,074个单核苷酸多态性(SNP)和7种模型,对200份亚麻核心种质进行了全基因组关联研究(GWAS)。共检测到434个与331个QTL相关的独特数量性状核苷酸(QTN)。在单倍型分析中发现,至少在两个数据集中共享的64个位点具有显著性,其中20个位点被多个模型共享。同时,重复检测到一个主效位点([位点名称缺失]),该位点存在于亚麻霜霉病抗性位点的定位研究中。油用亚麻比纤维用亚麻具有更多的正向效应或有利等位基因的QTL(PQTL),且对白粉病的抗性更高,表明这些QTL的效应主要是累加的。此外,鉴定出一个优良抗病品种C120,可用于推广种植。基于通过GWAS鉴定出的331个QTL和统计模型GBLUP,构建了一个与亚麻抗白粉病相关的基因组选择(GS)模型,预测准确率为0.96。我们的研究结果为抗性的遗传基础提供了有价值的见解,并有助于推进育种计划。