He Liqiang, Xiao Jin, Rashid Khalid Y, Yao Zhen, Li Pingchuan, Jia Gaofeng, Wang Xiue, Cloutier Sylvie, You Frank M
Ottawa Research and Development Centre, Agriculture and Agri-Food Canada, Ottawa, ON, Canada.
Key Laboratory of Crop Genetics and Germplasm Enhancement, College of Agriculture, Nanjing Agricultural University/JCIC-MCP, Nanjing, China.
Front Plant Sci. 2019 Jan 14;9:1982. doi: 10.3389/fpls.2018.01982. eCollection 2018.
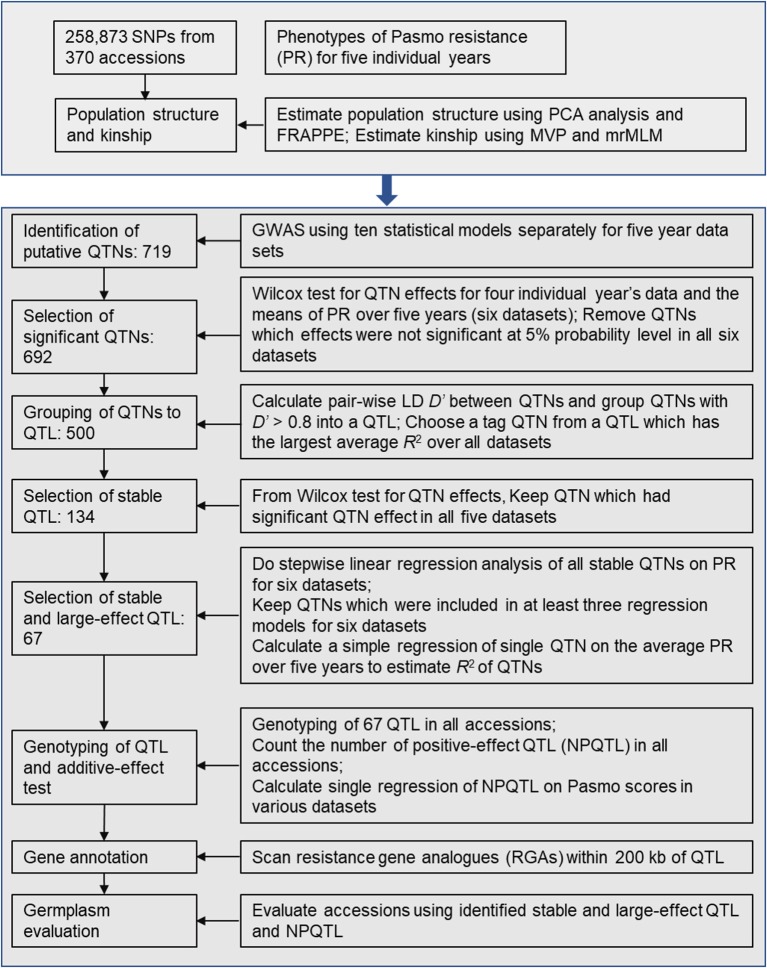
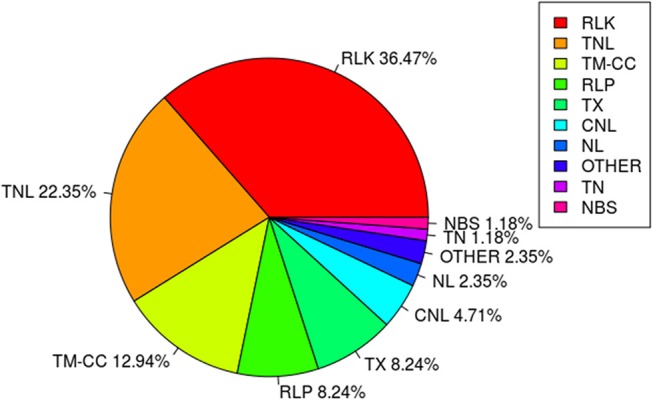
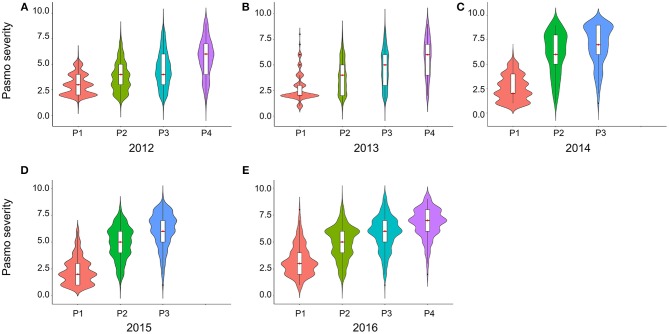
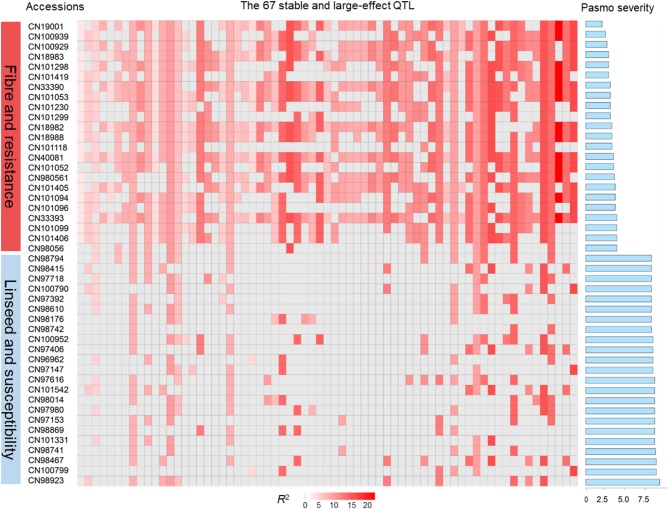
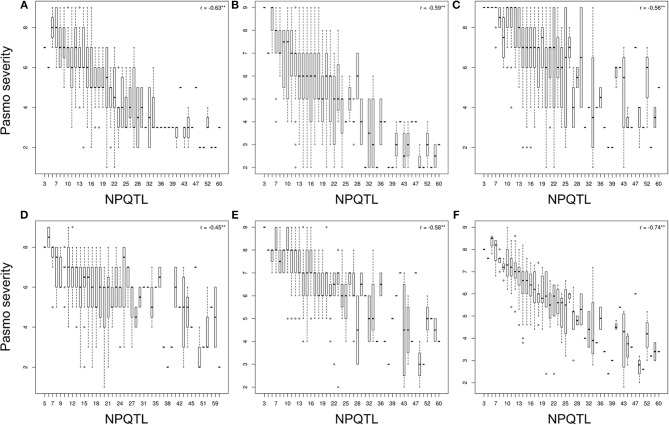
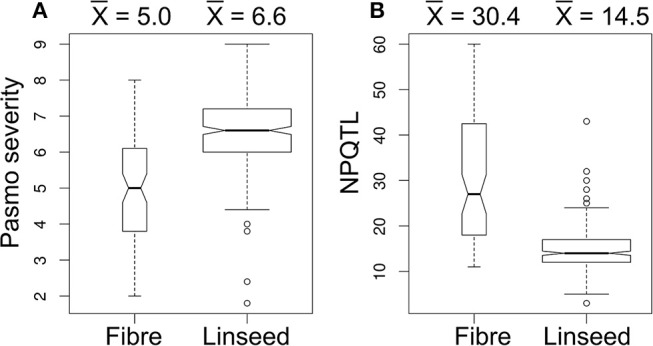
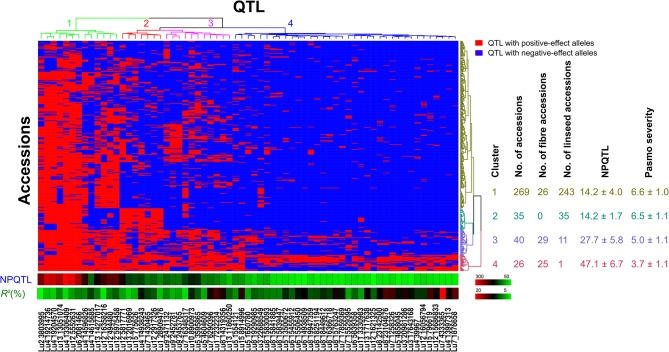
Pasmo is one of the most widespread diseases threatening flax production. To identify genetic regions associated with pasmo resistance (PR), a genome-wide association study was performed on 370 accessions from the flax core collection. Evaluation of pasmo severity was performed in the field from 2012 to 2016 in Morden, MB, Canada. Genotyping-by-sequencing has identified 258,873 single nucleotide polymorphisms (SNPs) distributed on all 15 flax chromosomes. Marker-trait associations were identified using ten different statistical models. A total of 692 unique quantitative trait nucleotides (QTNs) associated with 500 putative quantitative trait loci (QTL) were detected from six phenotypic PR datasets (five individual years and average across years). Different QTNs were identified with various statistical models and from individual PR datasets, indicative of the complementation between analytical methods and/or genotype × environment interactions of the QTL effects. The single-locus models tended to identify large-effect QTNs while the multi-loci models were able to detect QTNs with smaller effects. Among the putative QTL, 67 had large effects (3-23%), were stable across all datasets and explained 32-64% of the total variation for PR in the various datasets. Forty-five of these QTL spanned 85 resistance gene analogs including a large toll interleukin receptor, nucleotide-binding site, leucine-rich repeat (TNL) type gene cluster on chromosome 8. The number of QTL with positive-effect or favorite alleles (NPQTL) in accessions was significantly correlated with PR ( = 0.55), suggesting that these QTL effects are mainly additive. NPQTL was also significantly associated with morphotype ( = 0.52) and major QTL with positive effect alleles were present in the fiber type accessions. The 67 large effect QTL are suited for marker-assisted selection and the 500 QTL for effective genomic prediction in PR molecular breeding.
帕斯莫病是威胁亚麻生产的最广泛传播的病害之一。为了鉴定与抗帕斯莫病(PR)相关的基因区域,对来自亚麻核心种质库的370份材料进行了全基因组关联研究。2012年至2016年在加拿大曼尼托巴省莫登的田间对帕斯莫病严重程度进行了评估。简化基因组测序鉴定出分布在所有15条亚麻染色体上的258,873个单核苷酸多态性(SNP)。使用十种不同的统计模型鉴定标记-性状关联。从六个表型PR数据集(五个单一年份和多年平均值)中检测到总共692个与500个假定的数量性状基因座(QTL)相关的独特数量性状核苷酸(QTN)。不同的统计模型和各个PR数据集中鉴定出了不同的QTN,这表明分析方法之间的互补性和/或QTL效应的基因型×环境互作。单基因座模型倾向于鉴定出效应大的QTN,而多基因座模型能够检测到效应较小的QTN。在假定的QTL中,有67个具有较大效应(3%-23%),在所有数据集中都很稳定,并解释了各个数据集中PR总变异的32%-64%。其中45个QTL跨越了85个抗性基因类似物,包括位于8号染色体上的一个大型Toll样白细胞介素受体、核苷酸结合位点、富含亮氨酸重复序列(TNL)型基因簇。材料中具有正向效应或有利等位基因的QTL数量(NPQTL)与PR显著相关(r = 0.55),表明这些QTL效应主要是加性效应。NPQTL也与形态类型显著相关(r = 0.52),并且纤维类型材料中存在具有正向效应等位基因的主要QTL。这67个大效应QTL适用于标记辅助选择,而这500个QTL适用于PR分子育种中的有效基因组预测。