Bioinformatics and Systems Biology Program, University of California, San Diego, La Jolla, California, United States of America.
Department of Bioengineering, University of California, San Diego, La Jolla, California, United States of America.
PLoS Comput Biol. 2020 Mar 2;16(3):e1007608. doi: 10.1371/journal.pcbi.1007608. eCollection 2020 Mar.
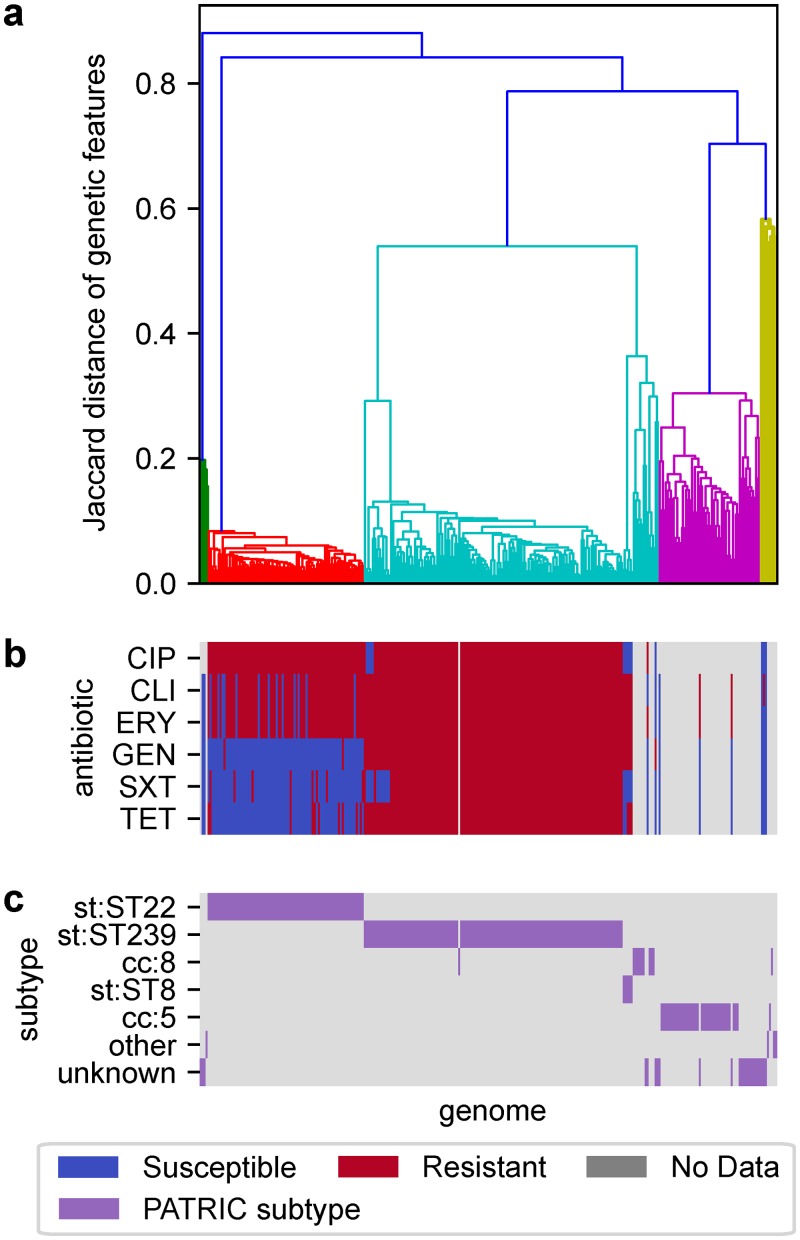
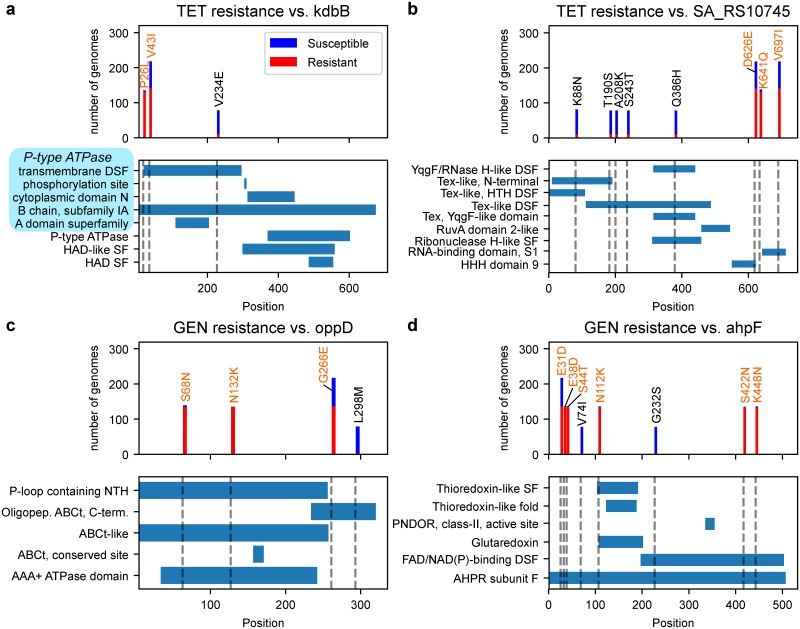
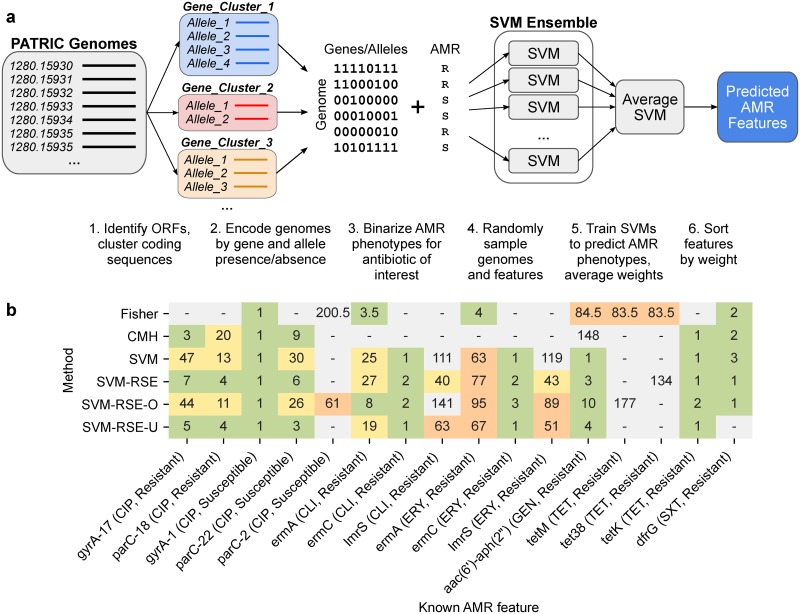
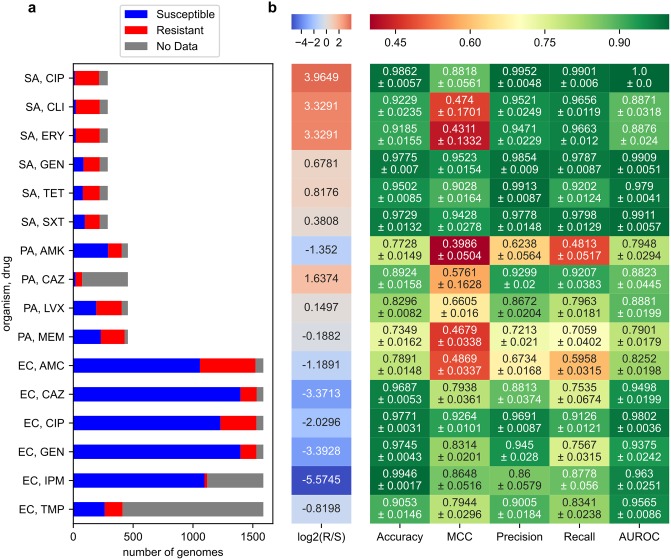
The evolution of antimicrobial resistance (AMR) poses a persistent threat to global public health. Sequencing efforts have already yielded genome sequences for thousands of resistant microbial isolates and require robust computational tools to systematically elucidate the genetic basis for AMR. Here, we present a generalizable machine learning workflow for identifying genetic features driving AMR based on constructing reference strain-agnostic pan-genomes and training random subspace ensembles (RSEs). This workflow was applied to the resistance profiles of 14 antimicrobials across three urgent threat pathogens encompassing 288 Staphylococcus aureus, 456 Pseudomonas aeruginosa, and 1588 Escherichia coli genomes. We find that feature selection by RSE detects known AMR associations more reliably than common statistical tests and previous ensemble approaches, identifying a total of 45 known AMR-conferring genes and alleles across the three organisms, as well as 25 candidate associations backed by domain-level annotations. Furthermore, we find that results from the RSE approach are consistent with existing understanding of fluoroquinolone (FQ) resistance due to mutations in the main drug targets, gyrA and parC, in all three organisms, and suggest the mutational landscape of those genes with respect to FQ resistance is simple. As larger datasets become available, we expect this approach to more reliably predict AMR determinants for a wider range of microbial pathogens.
抗菌药物耐药性(AMR)的进化对全球公共卫生构成持续威胁。测序工作已经产生了数千个耐药微生物分离株的基因组序列,需要强大的计算工具来系统阐明 AMR 的遗传基础。在这里,我们提出了一种基于构建参考菌株无关泛基因组和训练随机子空间集合(RSE)来识别驱动 AMR 的遗传特征的可推广机器学习工作流程。该工作流程应用于涵盖 288 个金黄色葡萄球菌、456 个铜绿假单胞菌和 1588 个大肠杆菌基因组的三种紧急威胁病原体的 14 种抗生素的耐药谱。我们发现,RSE 的特征选择比常见的统计检验和以前的集合方法更可靠地检测到已知的 AMR 关联,总共在这三种生物中鉴定出 45 个已知的赋予 AMR 的基因和等位基因,以及 25 个通过域级注释支持的候选关联。此外,我们发现 RSE 方法的结果与三种生物中主要药物靶标 gyrA 和 parC 突变导致氟喹诺酮(FQ)耐药的现有认识一致,并表明这些基因的 FQ 耐药突变景观是简单的。随着更大的数据集的出现,我们预计这种方法将更可靠地预测更广泛的微生物病原体的 AMR 决定因素。