Pathogen Biology Laboratory, Department of Biotechnology and Bioinformatics, University of Hyderabadgrid.18048.35, Hyderabad, India.
mBio. 2021 Feb 22;13(1):e0379621. doi: 10.1128/mbio.03796-21. Epub 2022 Feb 15.
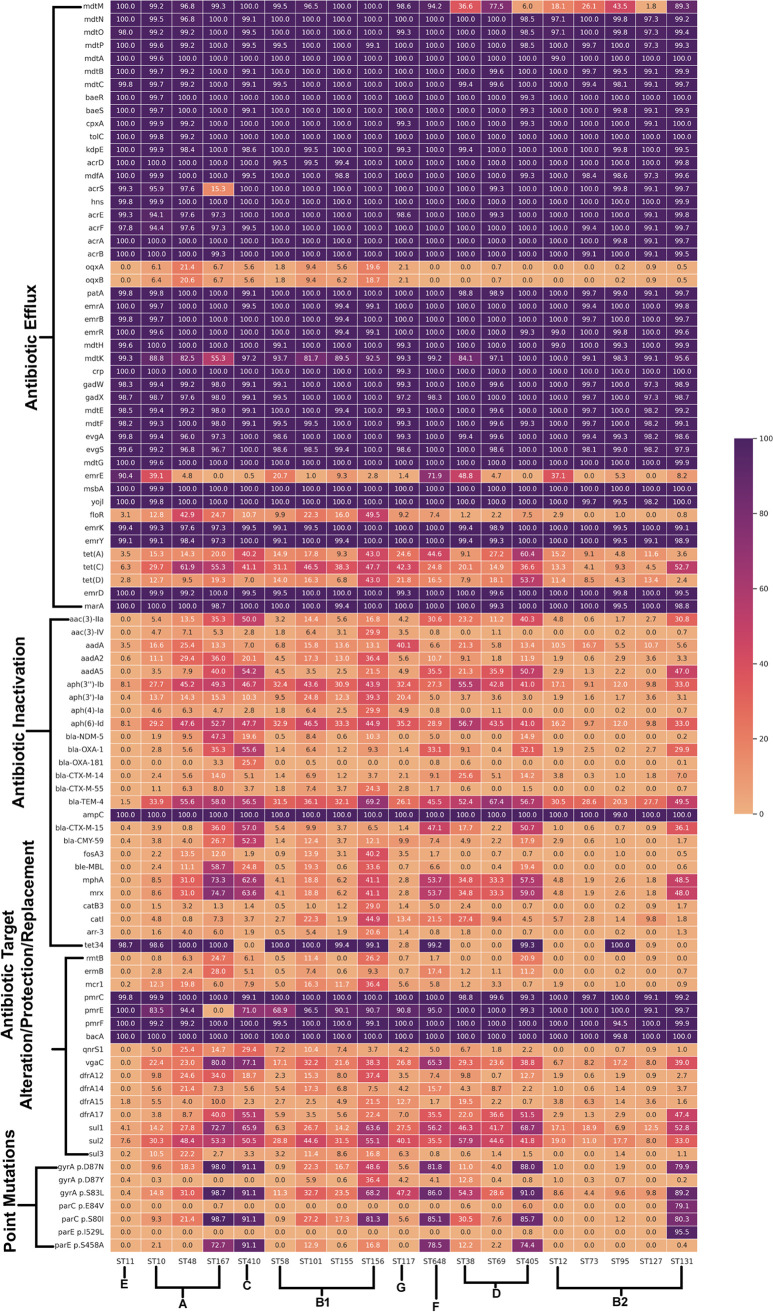
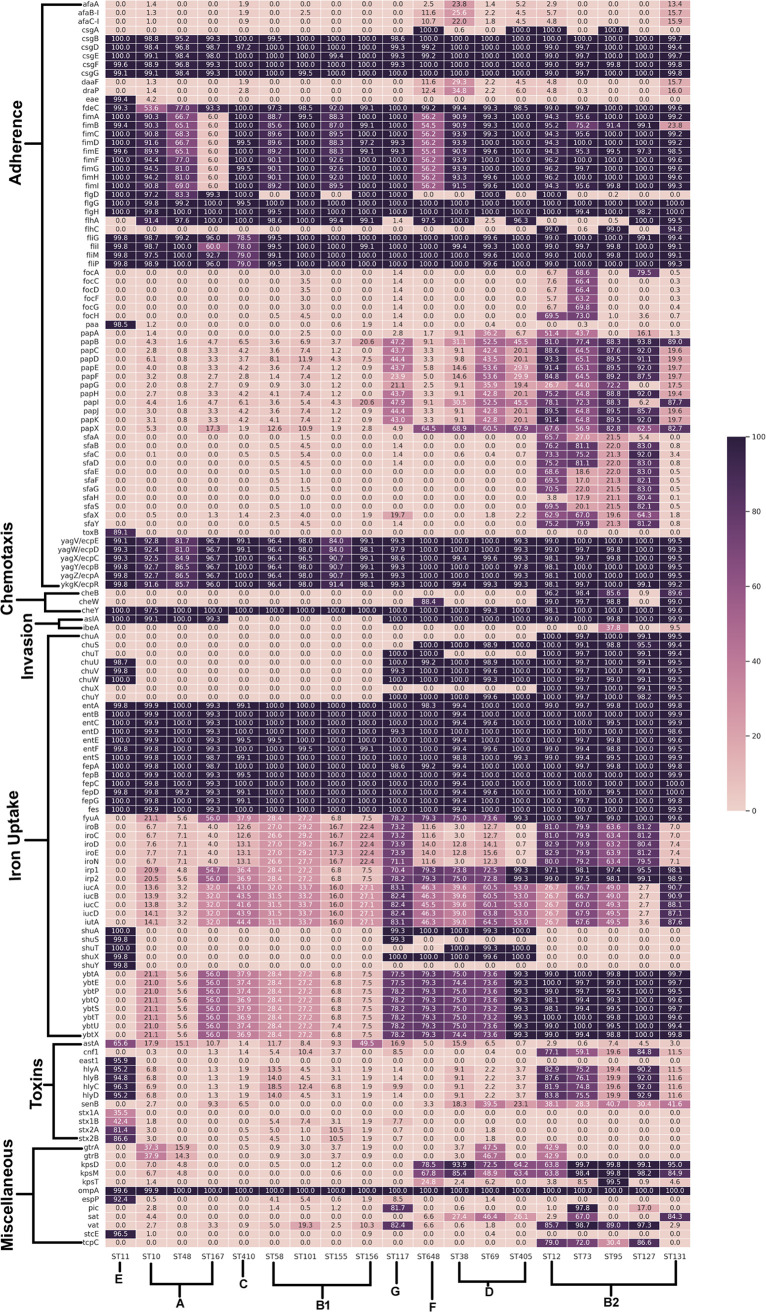
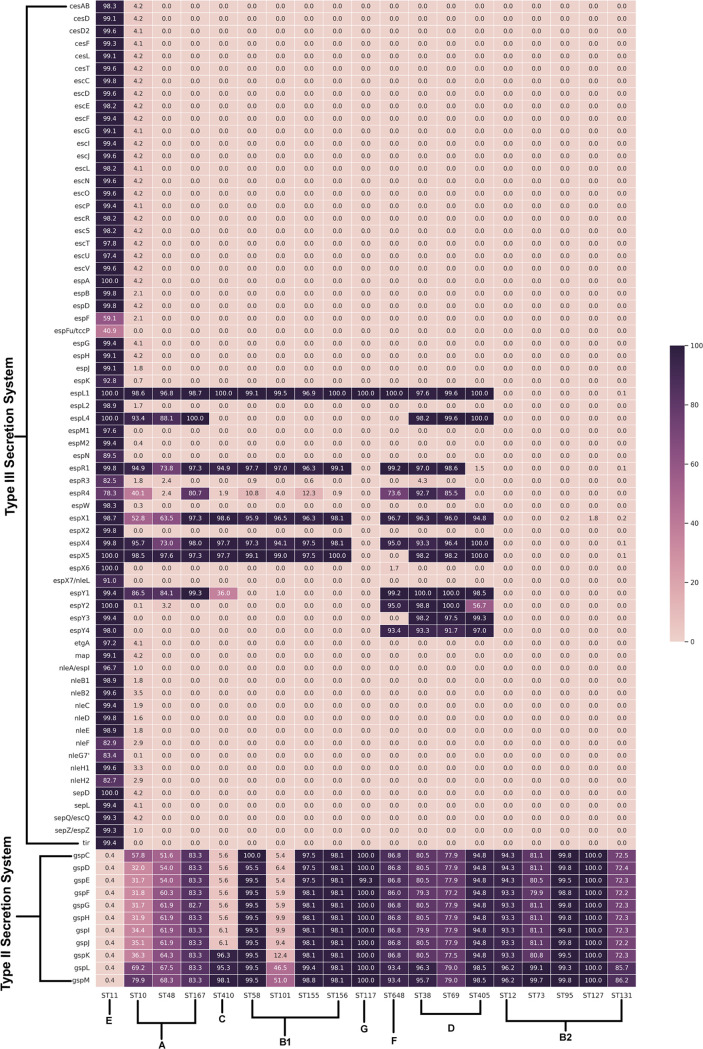
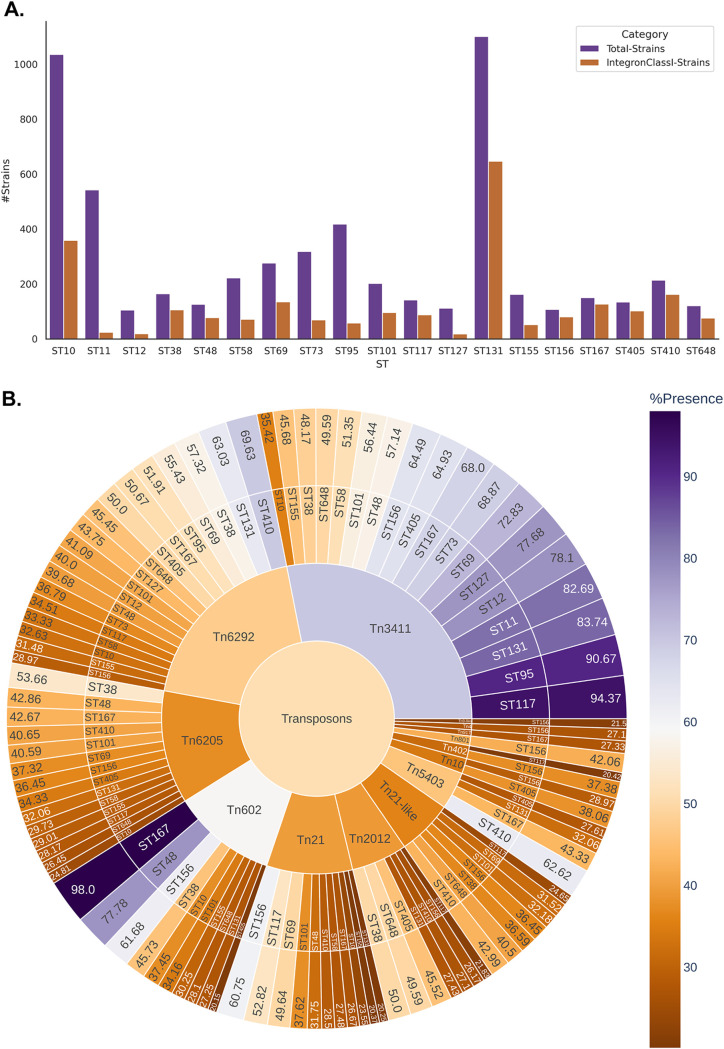
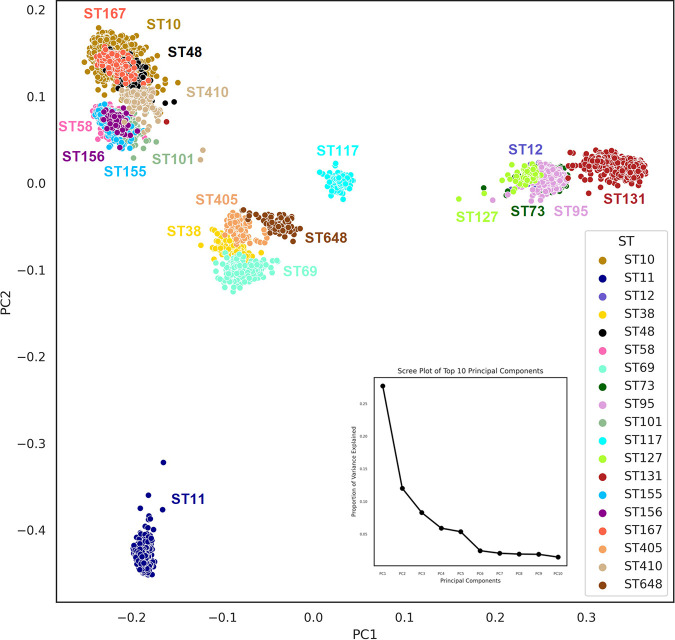
Escherichia coli, a ubiquitous commensal/pathogenic member from the family, accounts for high infection burden, morbidity, and mortality throughout the world. With emerging multidrug resistance (MDR) on a massive scale, E. coli has been listed as one of the Global Antimicrobial Resistance and Use Surveillance System (GLASS) priority pathogens. Understanding the resistance mechanisms and underlying genomic features appears to be of utmost importance to tackle further spread of these multidrug-resistant superbugs. While a few of the globally prevalent sequence types (STs) of E. coli, such as ST131, ST69, ST405, and ST648, have been previously reported to be highly virulent and harboring MDR, there is no clarity if certain ST lineages have a greater propensity to acquire MDR. In this study, large-scale comparative genomics of a total of 5,653 E. coli genomes from 19 ST lineages revealed ST-wide prevalence patterns of genomic features, such as antimicrobial resistance (AMR)-encoding genes/mutations, virulence genes, integrons, and transposons. Interpretation of the importance of these features using a Random Forest Classifier trained with 11,988 genomic features from whole-genome sequence data identified ST-specific or phylogroup-specific signature proteins mostly belonging to different protein superfamilies, including the toxin-antitoxin systems. Our study provides a comprehensive understanding of a myriad of genomic features, ST-specific proteins, and resistance mechanisms entailing different lineages of E. coli at the level of genomes; this could be of significant downstream importance in understanding the mechanisms of AMR, in clinical discovery, in epidemiology, and in devising control strategies. With the leap in whole-genome data being generated, the application of relevant methods to mine biologically significant information from microbial genomes is of utmost importance to public health genomics. Machine-learning methods have been used not only to mine, curate, or classify the data but also to identify the relevant features that could be linked to a particular class/target. This is perhaps one of the pioneering studies that has attempted to classify a large repertoire of E. coli genome data sets (5,653 genomes) belonging to 19 different STs (including well-studied as well as understudied STs) using machine learning approaches. Important features identified by these approaches have revealed ST-specific signature proteins, which could be further studied to predict possible associations with the phenotypic profiles, thereby providing a better understanding of virulence and the resistance mechanisms among different clonal lineages of E. coli.
大肠杆菌是家族中的一种普遍存在的共生/致病成员,在全球范围内造成了很高的感染负担、发病率和死亡率。随着大规模出现的多药耐药性(MDR),大肠杆菌已被列为全球抗菌药物耐药性和使用监测系统(GLASS)优先病原体之一。了解耐药机制和潜在的基因组特征对于遏制这些多药耐药超级细菌的进一步传播似乎至关重要。虽然一些全球流行的大肠杆菌序列类型(ST),如 ST131、ST69、ST405 和 ST648,以前被报道具有高度毒性和携带 MDR,但目前尚不清楚某些 ST 谱系是否更容易获得 MDR。在这项研究中,对来自 19 个 ST 谱系的总共 5653 个大肠杆菌基因组进行了大规模比较基因组学研究,揭示了基因组特征(如抗微生物药物耐药性(AMR)编码基因/突变、毒力基因、整合子和转座子)的 ST 广泛流行模式。使用随机森林分类器对来自全基因组序列数据的 11988 个基因组特征进行训练,对这些特征的重要性进行解释,确定了属于不同蛋白质超家族的 ST 特异性或进化枝特异性特征蛋白,包括毒素-抗毒素系统。我们的研究提供了对大肠杆菌不同谱系的基因组水平上的大量基因组特征、ST 特异性蛋白和耐药机制的全面了解;这对于理解 AMR 机制、临床发现、流行病学和制定控制策略可能具有重要的下游意义。随着全基因组数据的飞跃式增长,应用相关方法从微生物基因组中挖掘具有生物学意义的信息对于公共卫生基因组学至关重要。机器学习方法不仅用于挖掘、整理或分类数据,还用于识别可能与特定类别/目标相关的相关特征。这也许是一项开创性的研究之一,它试图使用机器学习方法对属于 19 个不同 ST(包括已研究和未研究的 ST)的大量大肠杆菌基因组数据集(5653 个基因组)进行分类。这些方法确定的重要特征揭示了 ST 特异性特征蛋白,这些蛋白可以进一步研究,以预测与表型谱的可能关联,从而更好地理解不同克隆谱系的大肠杆菌的毒力和耐药机制。