Laboratoire MIVEGEC (Université de Montpellier-CNRS-IRD), CREES, Montpellier, France.
Service de Parasitologie-mycologie CNR du Paludisme, AP-HP Hôpital Bichat, Paris, France.
PLoS Negl Trop Dis. 2020 Mar 9;14(3):e0008072. doi: 10.1371/journal.pntd.0008072. eCollection 2020 Mar.
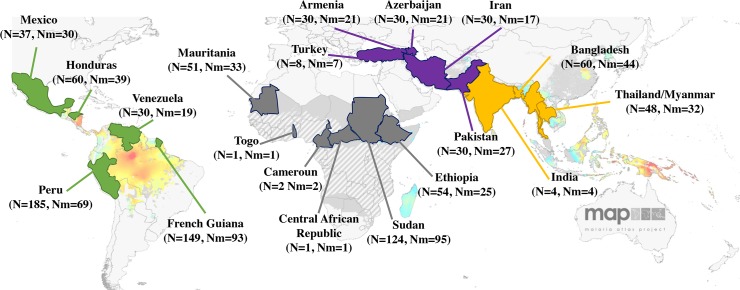
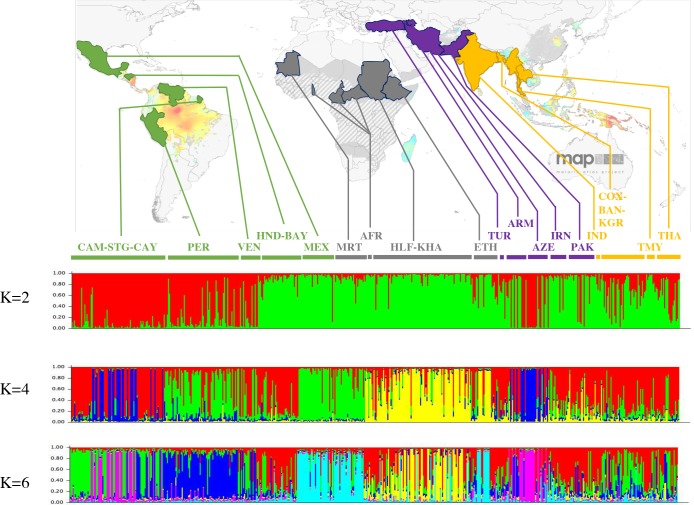
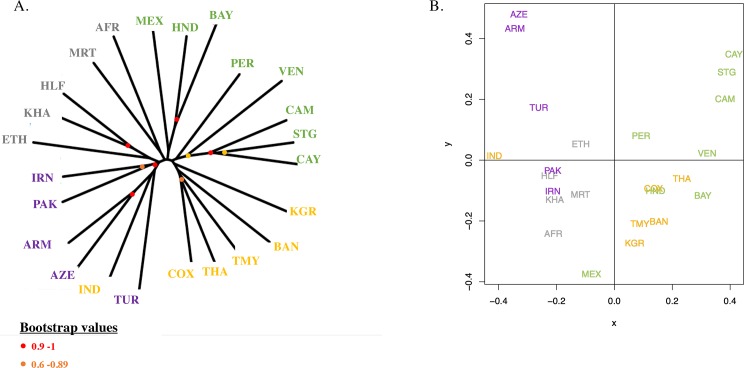
More than 200 million malaria clinical cases are reported each year due to Plasmodium vivax, the most widespread Plasmodium species in the world. This species has been neglected and understudied for a long time, due to its lower mortality in comparison with Plasmodium falciparum. A renewed interest has emerged in the past decade with the discovery of antimalarial drug resistance and of severe and even fatal human cases. Nonetheless, today there are still significant gaps in our understanding of the population genetics and evolutionary history of P. vivax, particularly because of a lack of genetic data from Africa. To address these gaps, we genotyped 14 microsatellite loci in 834 samples obtained from 28 locations in 20 countries from around the world. We discuss the worldwide population genetic structure and diversity and the evolutionary origin of P. vivax in the world and its introduction into the Americas. This study demonstrates the importance of conducting genome-wide analyses of P. vivax in order to unravel its complex evolutionary history.
每年有超过 2 亿例疟疾病例是由世界上分布最广的疟原虫——间日疟原虫引起的。由于间日疟原虫的死亡率相对较低,这种疟原虫长期以来一直被忽视和研究不足。在过去十年中,随着抗疟药物耐药性的发现以及严重甚至致命的人类病例的出现,人们对它的兴趣重新燃起。尽管如此,我们对间日疟原虫的群体遗传学和进化历史的了解仍然存在很大差距,特别是因为缺乏来自非洲的遗传数据。为了解决这些差距,我们对来自全球 20 个国家的 28 个地点的 834 个样本中的 14 个微卫星基因座进行了基因分型。我们讨论了间日疟原虫在全球的群体遗传结构和多样性,以及它在世界范围内的进化起源及其在美洲的传入。本研究表明,为了揭示间日疟原虫复杂的进化历史,对其进行全基因组分析非常重要。