Kaler Avjinder S, Gillman Jason D, Beissinger Timothy, Purcell Larry C
Department of Crop, Soil, and Environmental Sciences, University of Arkansas, Fayetteville, AR, United States.
Plant Genetic Research Unit, USDA-ARS, Columbia, MO, United States.
Front Plant Sci. 2020 Feb 25;10:1794. doi: 10.3389/fpls.2019.01794. eCollection 2019.
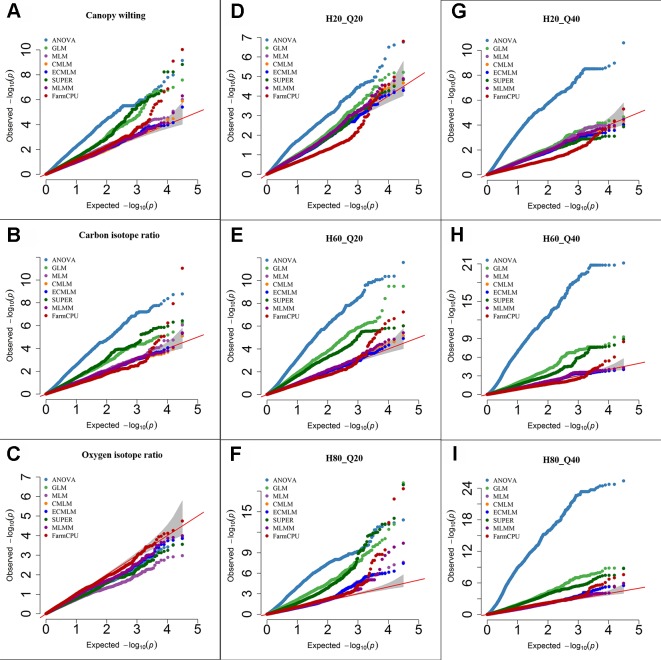
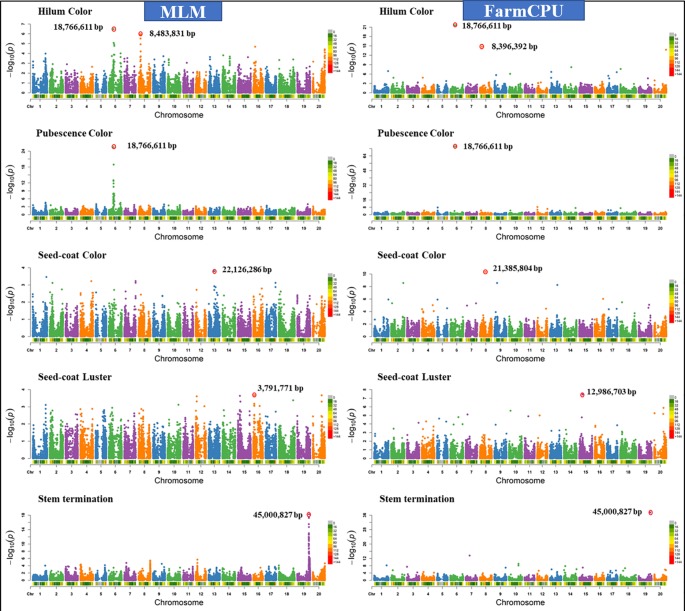
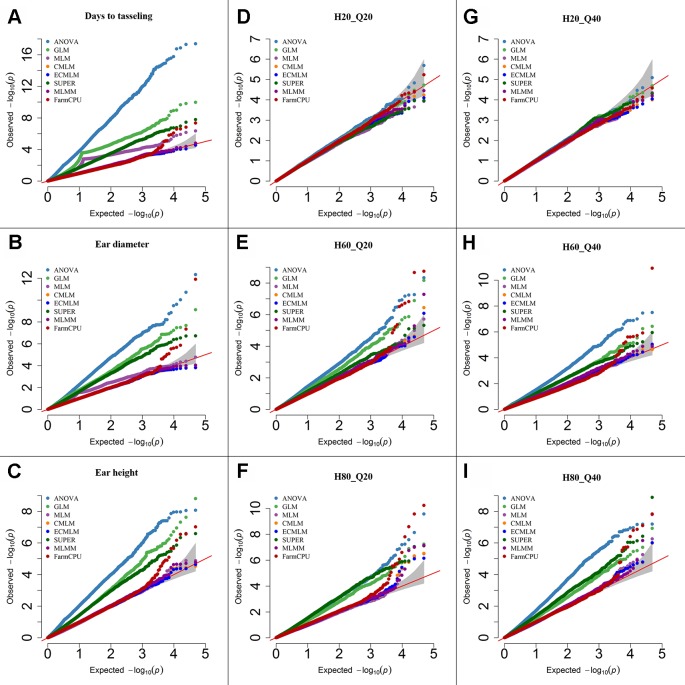
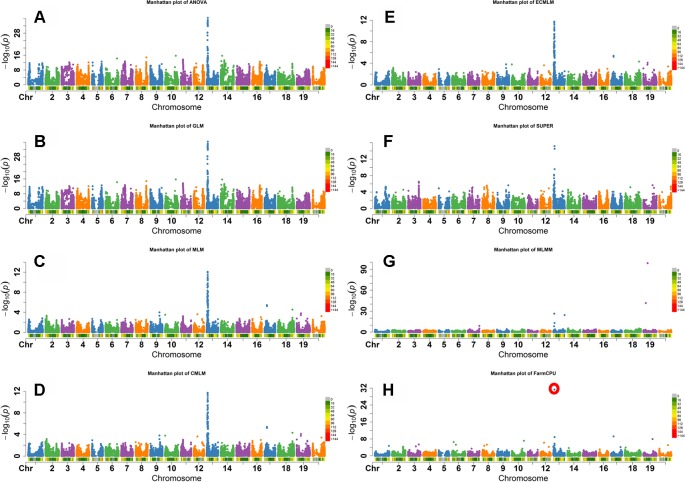
Association mapping (AM) is a powerful tool for fine mapping complex trait variation down to nucleotide sequences by exploiting historical recombination events. A major problem in AM is controlling false positives that can arise from population structure and family relatedness. False positives are often controlled by incorporating covariates for structure and kinship in mixed linear models (MLM). These MLM-based methods are single locus models and can introduce false negatives due to over fitting of the model. In this study, eight different statistical models, ranging from single-locus to multilocus, were compared for AM for three traits differing in heritability in two crop species: soybean ( L.) and maize ( L.). Soybean and maize were chosen, in part, due to their highly differentiated rate of linkage disequilibrium (LD) decay, which can influence false positive and false negative rates. The fixed and random model circulating probability unification (FarmCPU) performed better than other models based on an analysis of Q-Q plots and on the identification of the known number of quantitative trait loci (QTLs) in a simulated data set. These results indicate that the FarmCPU controls both false positives and false negatives. Six qualitative traits in soybean with known published genomic positions were also used to compare these models, and results indicated that the FarmCPU consistently identified a single highly significant SNP closest to these known published genes. Multiple comparison adjustments (Bonferroni, false discovery rate, and positive false discovery rate) were compared for these models using a simulated trait having 60% heritability and 20 QTLs. Multiple comparison adjustments were overly conservative for MLM, CMLM, ECMLM, and MLMM and did not find any significant markers; in contrast, ANOVA, GLM, and SUPER models found an excessive number of markers, far more than 20 QTLs. The FarmCPU model, using less conservative methods (false discovery rate, and positive false discovery rate) identified 10 QTLs, which was closer to the simulated number of QTLs than the number found by other models.
关联分析(AM)是一种强大的工具,可通过利用历史重组事件将复杂性状变异精细定位到核苷酸序列。AM中的一个主要问题是控制由群体结构和家族相关性引起的假阳性。通常通过在混合线性模型(MLM)中纳入结构和亲属关系的协变量来控制假阳性。这些基于MLM的方法是单基因座模型,由于模型过度拟合可能会引入假阴性。在本研究中,针对两种作物(大豆(Glycine max (L.) Merr.)和玉米(Zea mays L.))中遗传力不同的三个性状,比较了从单基因座到多基因座的八种不同统计模型用于AM。选择大豆和玉米部分原因是它们的连锁不平衡(LD)衰减速率高度不同,这会影响假阳性和假阴性率。基于Q-Q图分析以及在模拟数据集中对已知数量的数量性状基因座(QTL)的识别,固定和随机模型循环概率统一法(FarmCPU)比其他模型表现更好。这些结果表明FarmCPU能同时控制假阳性和假阴性。还使用了大豆中六个已知已发表基因组位置的质量性状来比较这些模型,结果表明FarmCPU始终能识别出最接近这些已知已发表基因的单个高度显著的SNP。使用具有60%遗传力和20个QTL的模拟性状,对这些模型的多重比较调整(邦费罗尼校正、错误发现率和阳性错误发现率)进行了比较。对于MLM、CMLM、ECMLM和MLMM,多重比较调整过于保守,未发现任何显著标记;相比之下,方差分析(ANOVA)、广义线性模型(GLM)和SUPER模型发现了过多的标记,远远超过20个QTL。使用不太保守方法(错误发现率和阳性错误发现率)的FarmCPU模型识别出10个QTL,比其他模型发现的数量更接近模拟的QTL数量。