Department of Reproduction Biology, National Institute of Medical Sciences and Nutrition Salvador Zubirán, Mexico City 14080, Mexico.
Tambre Foundation, Madrid 28002, Spain.
Asian J Androl. 2020 Nov-Dec;22(6):608-615. doi: 10.4103/aja.aja_143_19.
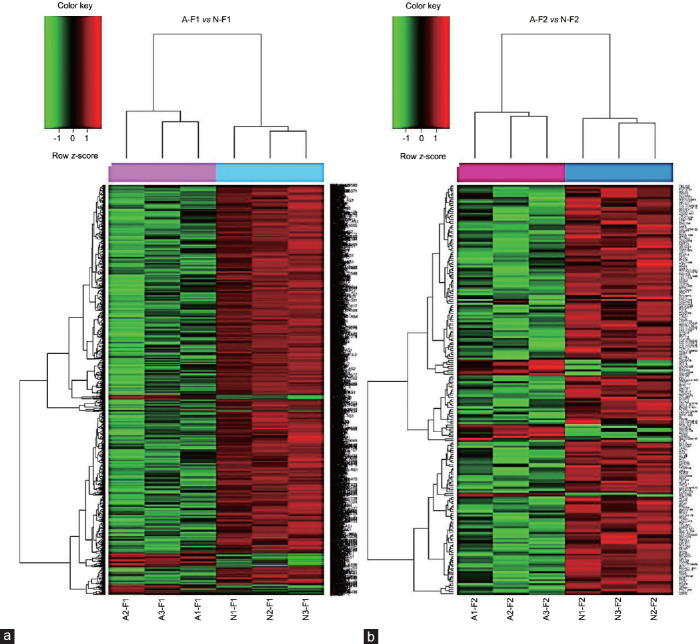
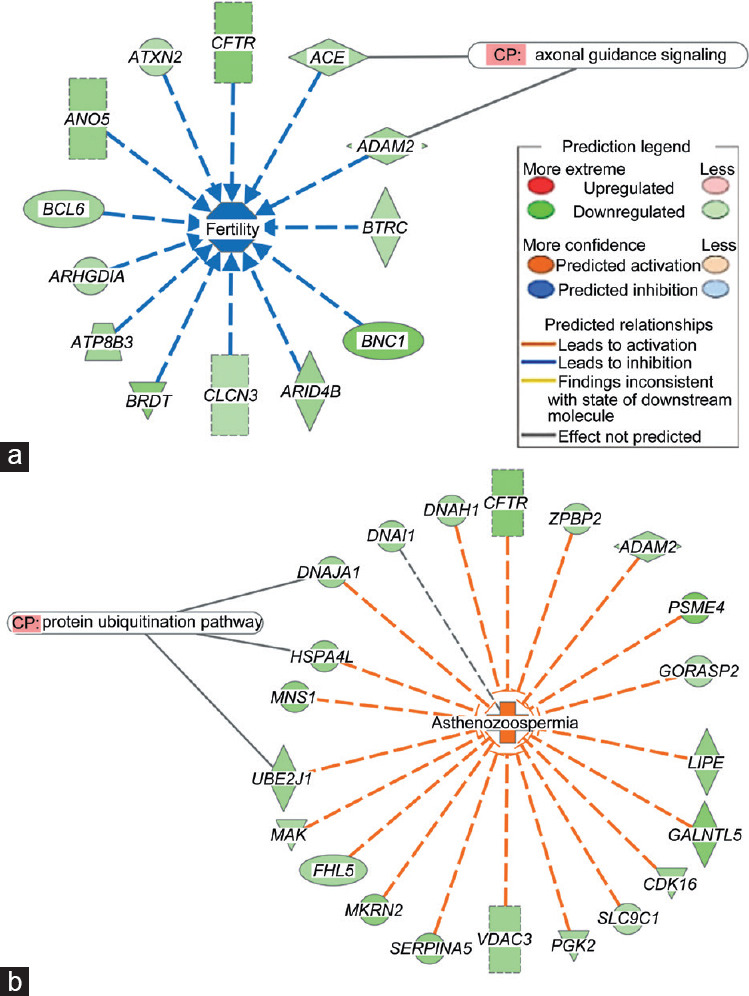
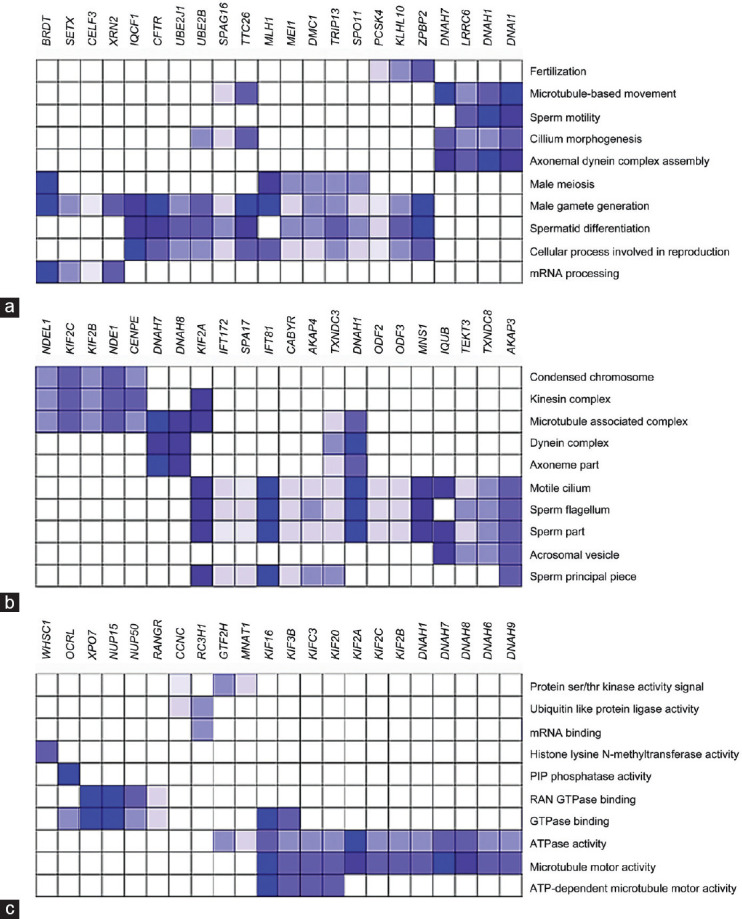
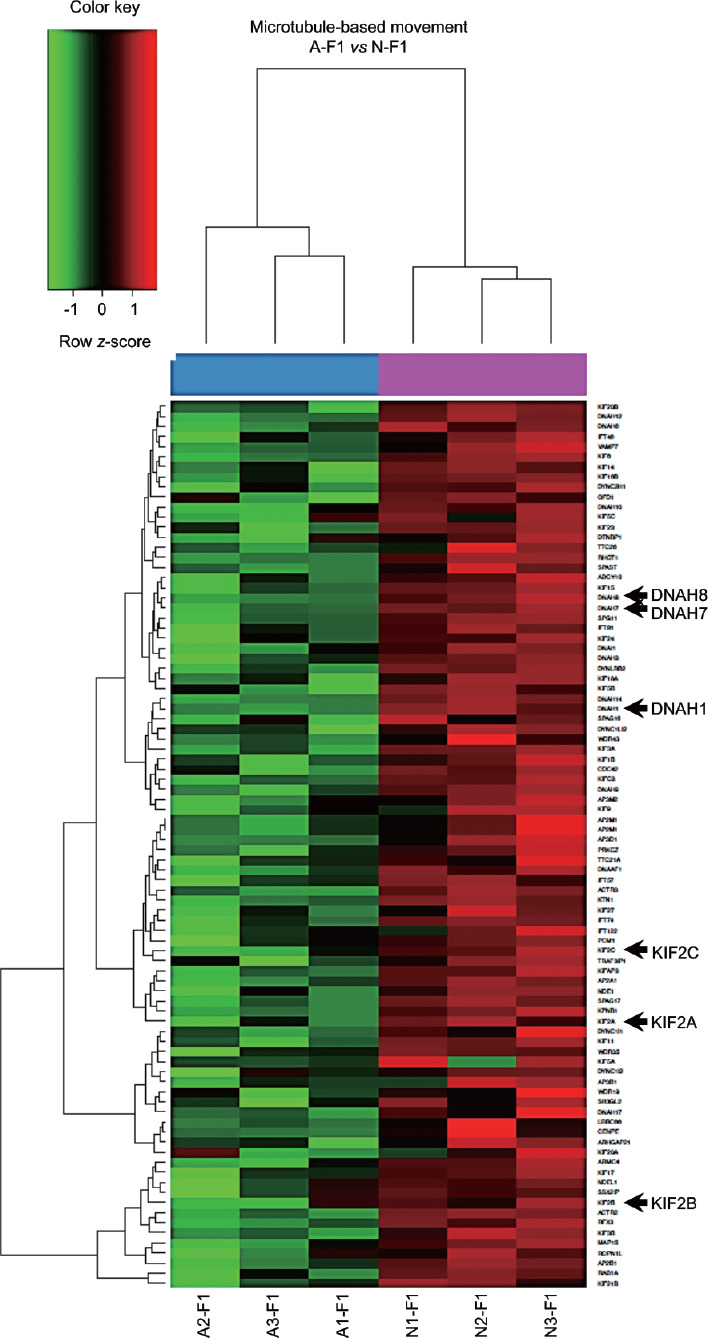
Spermatozoa contain a repertoire of RNAs considered to be potential functional fertility biomarkers. In this study, the gene expression of human sperm subpopulations with high (F1) and low (F2) motility from healthy normozoospermic (N) and asthenozoospermic (A) individuals was evaluated using RNA microarray followed by functional genomic analysis of differentially expressed genes. Results from A-F1 versus N-F1, A-F2 versus N-F2, N-F1 versus N-F2, and A-F1 versus A-F2 comparisons showed a considerably larger set of downregulated genes in tests versus controls. Gene ontology (GO) analysis of A-F1 versus N-F1 identified 507 overrepresented biological processes (BPs), several of which are associated with sperm physiology. In addition, gene set enrichment analysis of the same contrast showed 110 BPs, 36 cellular components, and 31 molecular functions, several of which are involved in sperm motility. A leading-edge analysis of selected GO terms resulted in several downregulated genes encoding to dyneins and kinesins, both related to sperm physiology. Furthermore, the predicted activation state of asthenozoospermia was increased, while fertility, cell movement of sperm, and gametogenesis were decreased. Interestingly, several downregulated genes characteristic of the canonical pathway protein ubiquitination were involved in asthenozoospermia activation. Conversely, GO analysis of A-F2 versus N-F2 did not identify overrepresented BPs, although the gene set enrichment analysis detected six enriched BPs, one cellular component, and two molecular functions. Overall, the results show differences in gene transcription between sperm subpopulations from asthenozoospermic and normozoospermic semen samples and allowed the identification of gene sets relevant to sperm physiology and reproduction.
精子包含一组被认为是潜在功能生育生物标志物的 RNA。在这项研究中,使用 RNA 微阵列评估了来自健康正常精子(N)和弱精子(A)个体的高(F1)和低(F2)活力精子亚群的基因表达,然后对差异表达基因进行功能基因组分析。A-F1 与 N-F1、A-F2 与 N-F2、N-F1 与 N-F2 和 A-F1 与 A-F2 比较的结果显示,测试组与对照组相比,下调基因的数量明显更多。A-F1 与 N-F1 比较的基因本体(GO)分析鉴定了 507 个过表达的生物学过程(BP),其中一些与精子生理学有关。此外,同一对比的基因集富集分析显示 110 个 BP、36 个细胞成分和 31 个分子功能,其中一些与精子运动有关。选定 GO 术语的前沿分析导致了几个下调基因的编码,这些基因编码与精子生理学相关的动力蛋白和驱动蛋白。此外,弱精子症的预测激活状态增加,而生育力、精子运动和配子发生减少。有趣的是,几个与经典途径蛋白泛素化相关的下调基因特征与弱精子症的激活有关。相反,A-F2 与 N-F2 比较的 GO 分析没有鉴定出过表达的 BP,尽管基因集富集分析检测到六个富集的 BP、一个细胞成分和两个分子功能。总的来说,这些结果显示了弱精子症和正常精子精液样本中精子亚群之间基因转录的差异,并鉴定了与精子生理学和生殖相关的基因集。