He Xibing, Liu Shuhan, Lee Tai-Sung, Ji Beihong, Man Viet H, York Darrin M, Wang Junmei
Department of Pharmaceutical Sciences and Computational Chemical Genomics Screening Center, School of Pharmacy, University of Pittsburgh, Pittsburgh, Pennsylvania 15261, United States.
Laboratory for Biomolecular Simulation Research, Center for Integrative Proteomics Research, and Department of Chemistry and Chemical Biology, Rutgers University, Piscataway, New Jersey 08854, United States.
ACS Omega. 2020 Feb 25;5(9):4611-4619. doi: 10.1021/acsomega.9b04233. eCollection 2020 Mar 10.
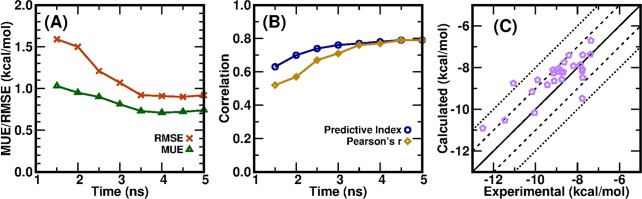
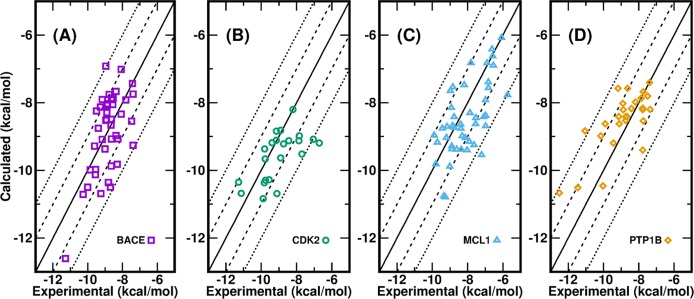
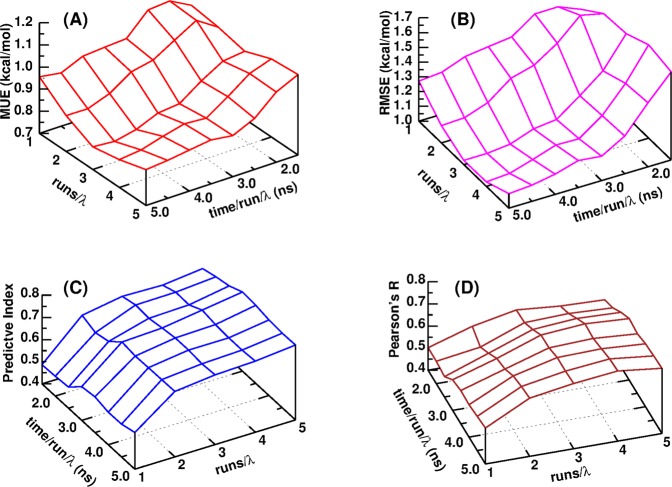
Accurate prediction of the absolute or relative protein-ligand binding affinity is one of the major tasks in computer-aided drug design projects, especially in the stage of lead optimization. In principle, the alchemical free energy (AFE) methods such as thermodynamic integration (TI) or free-energy perturbation (FEP) can fulfill this task, but in practice, a lot of hurdles prevent them from being routinely applied in daily drug design projects, such as the demanding computing resources, slow computing processes, unavailable or inaccurate force field parameters, and difficult and unfriendly setting up and post-analysis procedures. In this study, we have exploited practical protocols of applying the CPU (central processing unit)-TI and newly developed GPU (graphic processing unit)-TI modules and other tools in the AMBER software package, combined with ff14SB/GAFF1.8 force fields, to conduct efficient and accurate AFE calculations on protein-ligand binding free energies. We have tested 134 protein-ligand complexes in total for four target proteins (BACE, CDK2, MCL1, and PTP1B) and obtained overall comparable performance with the commercial Schrodinger FEP+ program (WangJ. Am. Chem. Soc.2015, 137, 2695-2703). The achieved accuracy fits within the requirements for computations to generate effective guidance for experimental work in drug lead optimization, and the needed wall time is short enough for practical application. Our verified protocol provides a practical solution for routine AFE calculations in real drug design projects.
准确预测蛋白质 - 配体的绝对或相对结合亲和力是计算机辅助药物设计项目中的主要任务之一,尤其是在先导化合物优化阶段。原则上,诸如热力学积分(TI)或自由能微扰(FEP)等炼金术自由能(AFE)方法可以完成此任务,但在实践中,许多障碍阻碍了它们在日常药物设计项目中的常规应用,例如所需的计算资源苛刻、计算过程缓慢、力场参数不可用或不准确,以及设置和后分析程序困难且不友好。在本研究中,我们利用了在AMBER软件包中应用CPU(中央处理器) - TI和新开发的GPU(图形处理器) - TI模块及其他工具的实用方案,并结合ff14SB/GAFF1.8力场,对蛋白质 - 配体结合自由能进行高效且准确的AFE计算。我们总共测试了针对四种靶蛋白(BACE、CDK2、MCL1和PTP1B)的134个蛋白质 - 配体复合物,并获得了与商业Schrodinger FEP+程序总体相当的性能(WangJ. Am. Chem. Soc.2015, 137, 2695 - 2703)。所达到的精度符合为药物先导优化实验工作生成有效指导的计算要求,并且所需的计算时间短到足以用于实际应用。我们经过验证的方案为实际药物设计项目中的常规AFE计算提供了一个实用的解决方案。