Population Health Sciences Institute, Faculty of Medical Sciences, Newcastle University, Newcastle upon Tyne, UK.
Division of Musculoskeletal and Dermatological Sciences, University of Manchester, Manchester, UK.
Genet Epidemiol. 2020 Jul;44(5):425-441. doi: 10.1002/gepi.22290. Epub 2020 Mar 19.
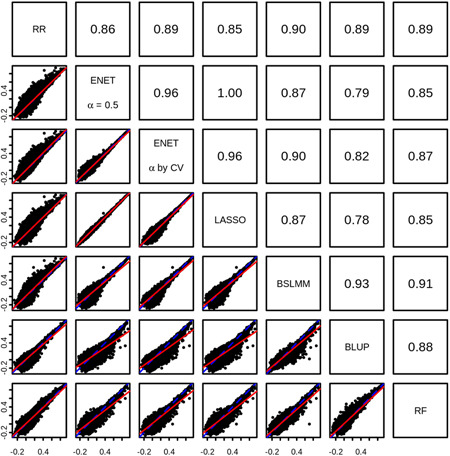
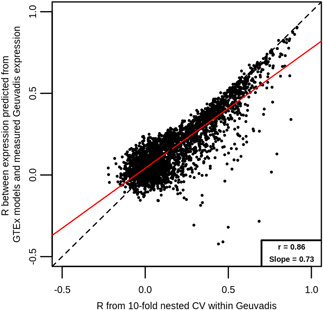
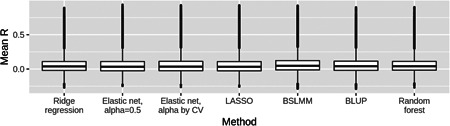
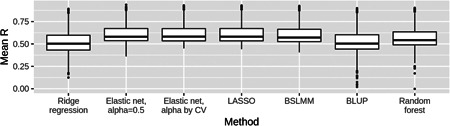
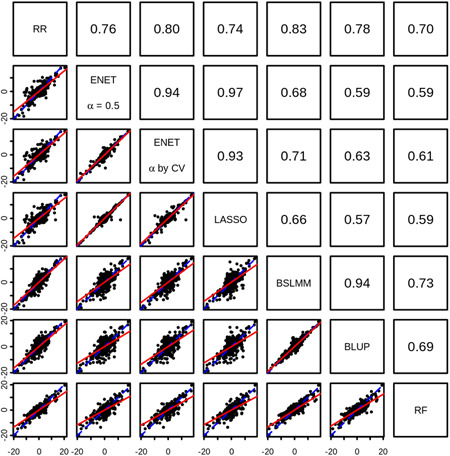
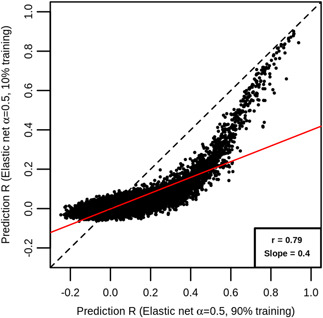
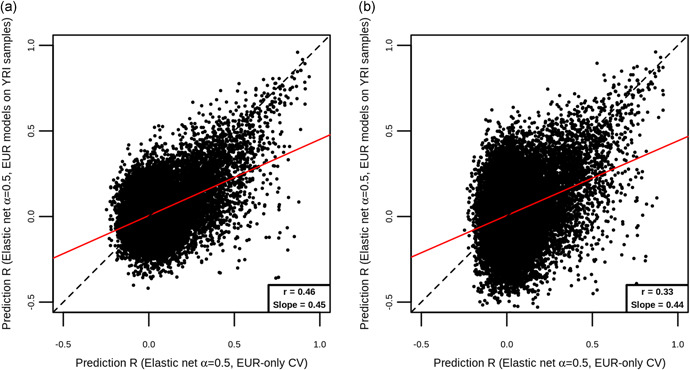
In transcriptome-wide association studies (TWAS), gene expression values are predicted using genotype data and tested for association with a phenotype. The power of this approach to detect associations relies, at least in part, on the accuracy of the prediction. Here we compare the prediction accuracy of six different methods-LASSO, Ridge regression, Elastic net, Best Linear Unbiased Predictor, Bayesian Sparse Linear Mixed Model, and Random Forests-by performing cross-validation using data from the Geuvadis Project. We also examine prediction accuracy (a) at different sample sizes, (b) when ancestry of the prediction model training and testing populations is different, and (c) when the tissue used to train the model is different from the tissue to be predicted. We find that, for most genes, the expression cannot be accurately predicted, but in general sparse statistical models tend to outperform polygenic models at prediction. Average prediction accuracy is reduced when the model training set size is reduced or when predicting across ancestries and is marginally reduced when predicting across tissues. We conclude that using sparse statistical models and the development of large reference panels across multiple ethnicities and tissues will lead to better prediction of gene expression, and thus may improve TWAS power.
在转录组全基因组关联研究(TWAS)中,使用基因型数据预测基因表达值,并测试其与表型的关联。该方法检测关联的能力至少部分依赖于预测的准确性。在这里,我们通过使用 Geuvadis 项目的数据进行交叉验证,比较了六种不同方法(LASSO、岭回归、弹性网络、最佳线性无偏预测、贝叶斯稀疏线性混合模型和随机森林)的预测准确性。我们还研究了预测准确性(a)在不同样本大小下的表现,(b)在预测模型训练和测试人群的祖源不同时的表现,以及(c)在用于训练模型的组织与要预测的组织不同时的表现。我们发现,对于大多数基因,表达不能被准确预测,但一般来说,稀疏统计模型在预测方面往往优于多基因模型。当模型训练集的大小减小时,平均预测准确性会降低,当跨祖源预测时,预测准确性会略有降低,当跨组织预测时,预测准确性也会略有降低。我们得出结论,使用稀疏统计模型和在多个种族和组织中开发大型参考面板将导致更好的基因表达预测,从而可能提高 TWAS 的能力。