School of Sport, Exercise and Rehabilitation Sciences, University of Birmingham, Birmingham, UK.
Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, UK.
FASEB J. 2020 May;34(5):6284-6301. doi: 10.1096/fj.201903051R. Epub 2020 Mar 22.
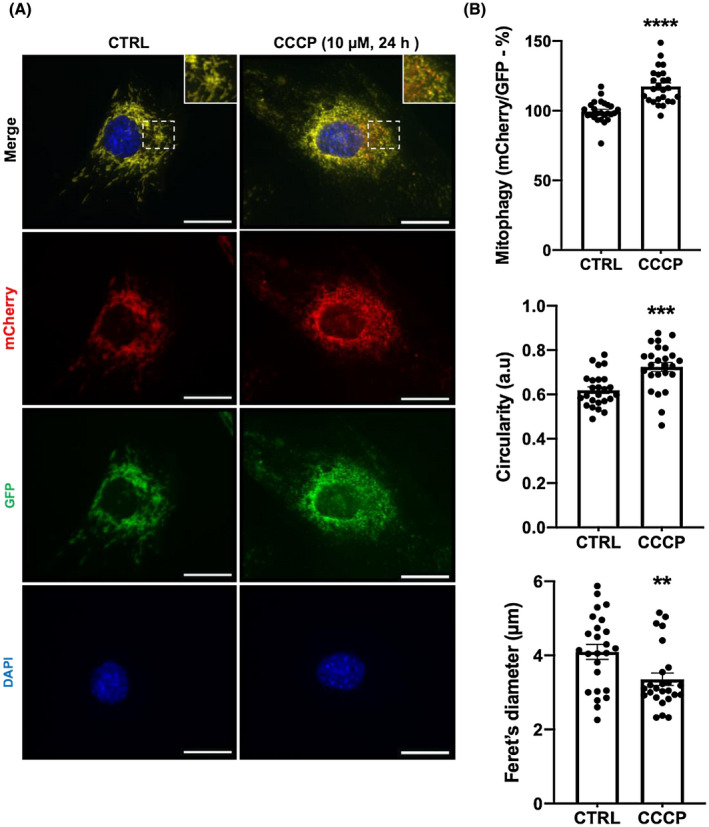
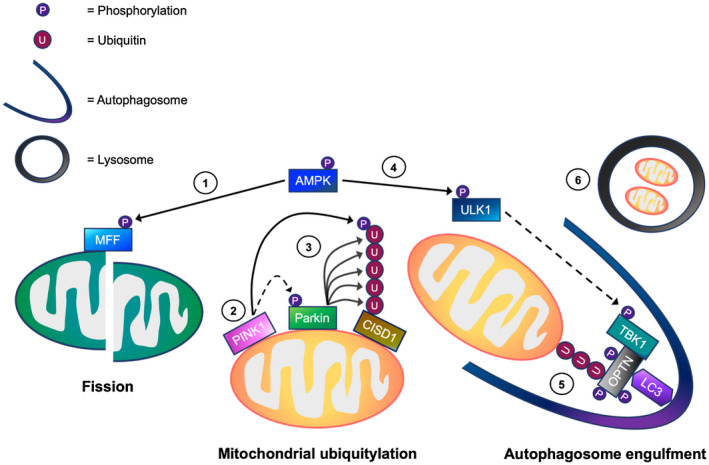
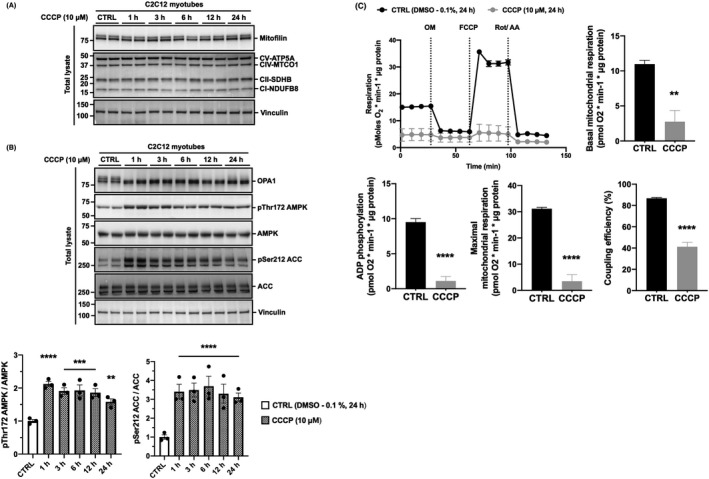
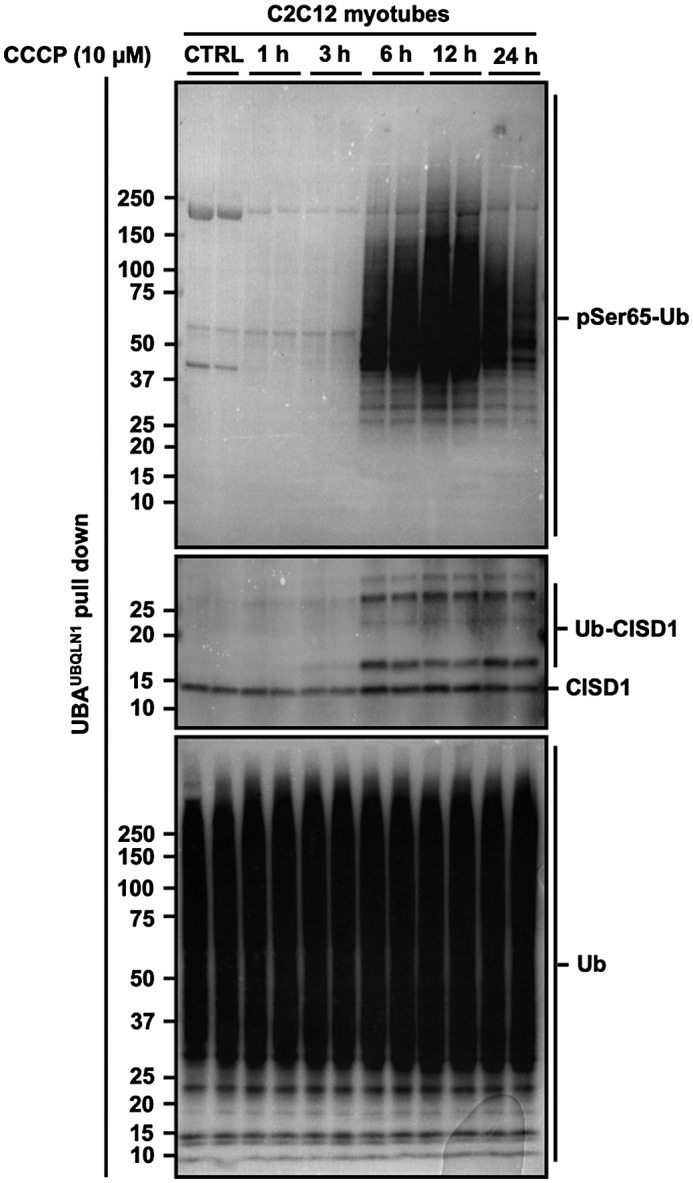
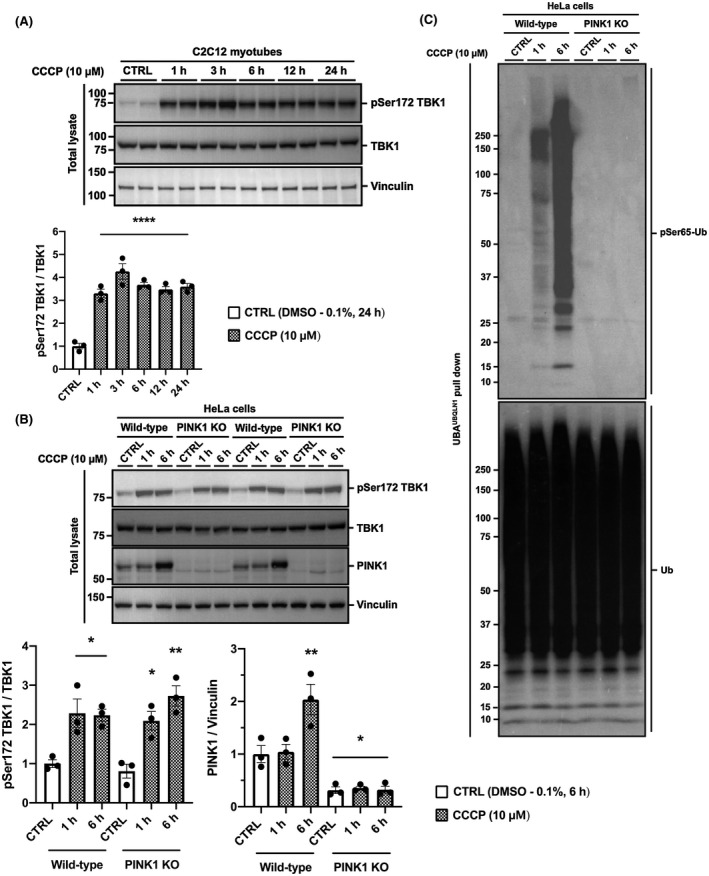
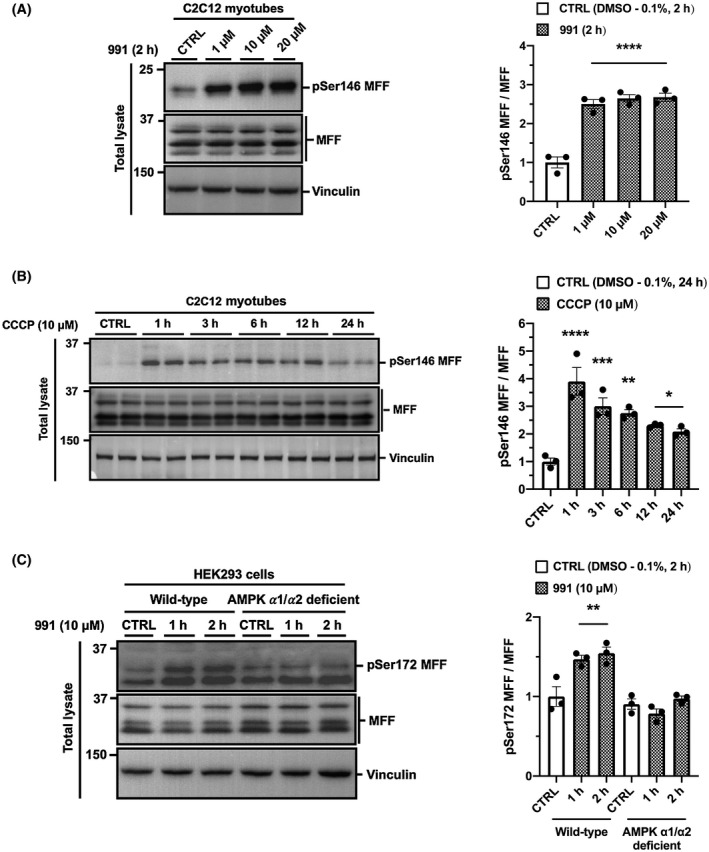
Mitophagy is a key process regulating mitochondrial quality control. Several mechanisms have been proposed to regulate mitophagy, but these have mostly been studied using stably expressed non-native proteins in immortalized cell lines. In skeletal muscle, mitophagy and its molecular mechanisms require more thorough investigation. To measure mitophagy directly, we generated a stable skeletal muscle C2C12 cell line, expressing a mitophagy reporter construct (mCherry-green fluorescence protein-mtFIS1 ). Here, we report that both carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment and adenosine monophosphate activated protein kinase (AMPK) activation by 991 promote mitochondrial fission via phosphorylation of MFF and induce mitophagy by ~20%. Upon CCCP treatment, but not 991, ubiquitin phosphorylation, a read-out of PTEN-induced kinase 1 (PINK1) activity, and Parkin E3 ligase activity toward CDGSH iron sulfur domain 1 (CISD1) were increased. Although the PINK1-Parkin signaling pathway is active in response to CCCP treatment, we observed no change in markers of mitochondrial protein content. Interestingly, our data shows that TANK-binding kinase 1 (TBK1) phosphorylation is increased after both CCCP and 991 treatments, suggesting TBK1 activation to be independent of both PINK1 and Parkin. Finally, we confirmed in non-muscle cell lines that TBK1 phosphorylation occurs in the absence of PINK1 and is regulated by AMPK-dependent signaling. Thus, AMPK activation promotes mitophagy by enhancing mitochondrial fission (via MFF phosphorylation) and autophagosomal engulfment (via TBK1 activation) in a PINK1-Parkin independent manner.
自噬是调节线粒体质量控制的关键过程。已经提出了几种调节自噬的机制,但这些机制大多是在用稳定表达的非天然蛋白在永生化细胞系中进行研究。在骨骼肌中,自噬及其分子机制需要更深入的研究。为了直接测量自噬,我们生成了一种稳定的骨骼肌 C2C12 细胞系,表达自噬报告构建体(mCherry-绿色荧光蛋白-mtFIS1)。在这里,我们报告说,羰基氰化物 m-氯代苯腙(CCCP)处理和腺苷单磷酸激活蛋白激酶(AMPK)通过磷酸化 MFF 激活(991)均可促进线粒体裂变,并通过 ~20%诱导自噬。在 CCCP 处理后,但不是 991 处理后,泛素磷酸化(PTEN 诱导激酶 1(PINK1)活性的读码器)和 Parkin E3 连接酶对 CDGSH 铁硫域 1(CISD1)的活性增加。尽管 PINK1-Parkin 信号通路对 CCCP 处理有反应,但我们观察到线粒体蛋白含量的标记物没有变化。有趣的是,我们的数据表明,在 CCCP 和 991 处理后,TANK 结合激酶 1(TBK1)磷酸化增加,表明 TBK1 激活独立于 PINK1 和 Parkin。最后,我们在非肌肉细胞系中证实,TBK1 磷酸化发生在没有 PINK1 的情况下,并且受 AMPK 依赖性信号调节。因此,AMPK 激活通过增强线粒体裂变(通过 MFF 磷酸化)和自噬体吞噬(通过 TBK1 激活),以一种不依赖于 PINK1 和 Parkin 的方式促进自噬。