Seabright Alex P, Lai Yu-Chiang
School of Sport, Exercise and Rehabilitation Sciences, University of Birmingham, Birmingham, United Kingdom.
Institute of Metabolism and Systems Research, University of Birmingham, Birmingham, United Kingdom.
Front Physiol. 2020 Dec 3;11:608474. doi: 10.3389/fphys.2020.608474. eCollection 2020.
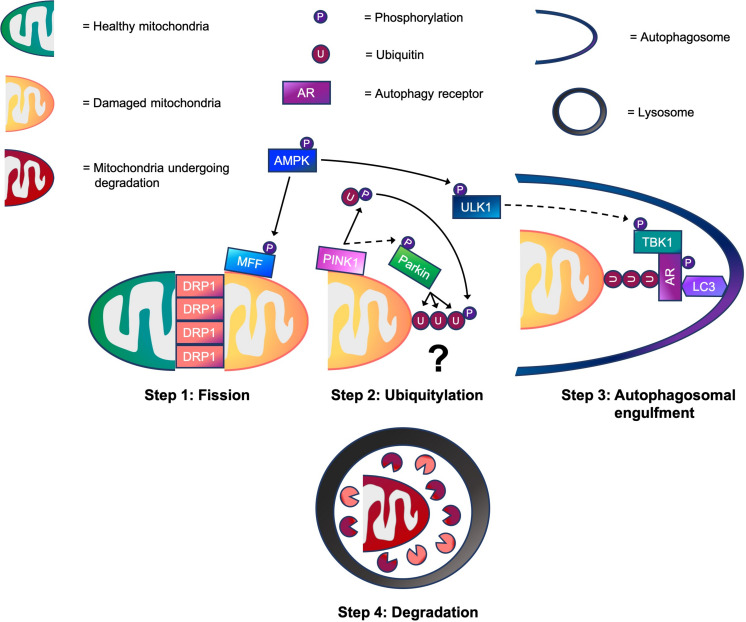
The selective removal of damaged mitochondria, also known as mitophagy, is an important mechanism that regulates mitochondrial quality control. Evidence suggests that mitophagy is adversely affected in aged skeletal muscle, and this is thought to contribute toward the age-related decline of muscle health. While our knowledge of the molecular mechanisms that regulate mitophagy are derived mostly from work in non-muscle cells, whether these mechanisms are conferred in muscle under physiological conditions has not been thoroughly investigated. Recent findings from our laboratory and those of others have made several novel contributions to this field. Herein, we consolidate current literature, including our recent work, while evaluating how ubiquitin-dependent mitophagy is regulated both in muscle and non-muscle cells through the steps of mitochondrial fission, ubiquitylation, and autophagosomal engulfment. During ubiquitin-dependent mitophagy in non-muscle cells, mitochondrial depolarization activates PINK1-Parkin signaling to elicit mitochondrial ubiquitylation. TANK-binding kinase 1 (TBK1) then activates autophagy receptors, which in turn, tether ubiquitylated mitochondria to autophagosomes prior to lysosomal degradation. In skeletal muscle, evidence supporting the involvement of PINK1-Parkin signaling in mitophagy is lacking. Instead, 5'-AMP-activated protein kinase (AMPK) is emerging as a critical regulator. Mechanistically, AMPK activation promotes mitochondrial fission before enhancing autophagosomal engulfment of damaged mitochondria possibly via TBK1. While TBK1 may be a point of convergence between PINK1-Parkin and AMPK signaling in muscle, the critical question that remains is: whether mitochondrial ubiquitylation is required for mitophagy. In future, improving understanding of molecular processes that regulate mitophagy in muscle will help to develop novel strategies to promote healthy aging.
选择性清除受损线粒体,即线粒体自噬,是调节线粒体质量控制的重要机制。有证据表明,线粒体自噬在老年骨骼肌中受到不利影响,这被认为是导致与年龄相关的肌肉健康衰退的原因之一。虽然我们对调节线粒体自噬的分子机制的了解大多来自非肌肉细胞的研究,但这些机制在生理条件下的肌肉中是否存在尚未得到充分研究。我们实验室和其他实验室的最新发现为该领域做出了一些新的贡献。在此,我们整合了当前的文献,包括我们最近的工作,同时评估了泛素依赖性线粒体自噬在肌肉和非肌肉细胞中是如何通过线粒体分裂、泛素化和自噬体吞噬等步骤进行调节的。在非肌肉细胞的泛素依赖性线粒体自噬过程中,线粒体去极化激活PINK1-Parkin信号通路,引发线粒体泛素化。然后,TANK结合激酶1(TBK1)激活自噬受体,自噬受体进而将泛素化的线粒体拴系到自噬体上,然后进行溶酶体降解。在骨骼肌中,缺乏支持PINK1-Parkin信号通路参与线粒体自噬的证据。相反,5'-AMP激活蛋白激酶(AMPK)正在成为一个关键的调节因子。从机制上讲,AMPK激活促进线粒体分裂,然后可能通过TBK1增强对受损线粒体的自噬体吞噬。虽然TBK1可能是肌肉中PINK1-Parkin和AMPK信号通路的交汇点,但仍然存在的关键问题是:线粒体自噬是否需要线粒体泛素化。未来,深入了解调节肌肉中线粒体自噬的分子过程将有助于开发促进健康衰老的新策略。