Microsoft Genomics Team, Redmond, WA, USA 98052.
Virginia Tech University, Dep. of Computer Science, Blacksburg, VA 24061, USA.
Biomed Res Int. 2020 Feb 25;2020:8531502. doi: 10.1155/2020/8531502. eCollection 2020.
Next-generation sequencing enables massively parallel processing, allowing lower cost than the other sequencing technologies. In the subsequent analysis with the NGS data, one of the major concerns is the reliability of variant calls. Although researchers can utilize raw quality scores of variant calling, they are forced to start the further analysis without any preevaluation of the quality scores.
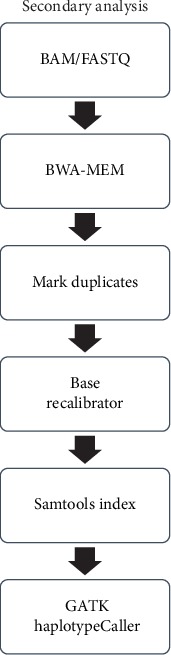
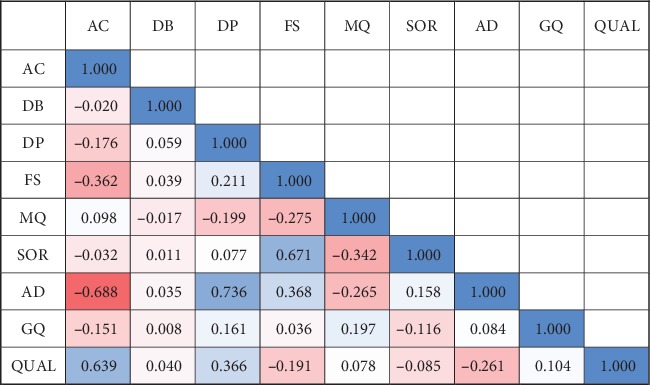
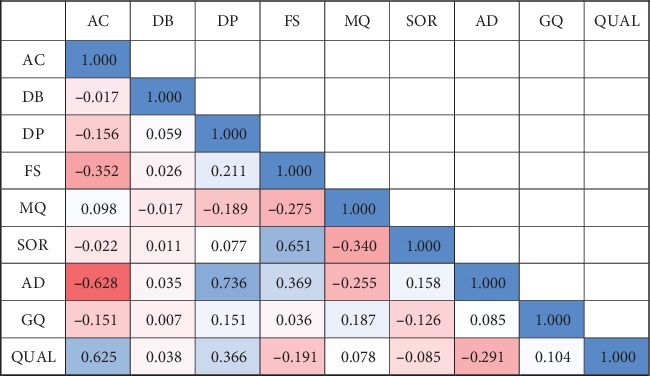
We presented a machine learning approach for estimating quality scores of variant calls derived from BWA+GATK. We analyzed correlations between the quality score and these annotations, specifying informative annotations which were used as features to predict variant quality scores. To test the predictive models, we simulated 24 paired-end Illumina sequencing reads with 30x coverage base. Also, twenty-four human genome sequencing reads resulting from Illumina paired-end sequencing with at least 30x coverage were secured from the Sequence Read Archive.
Using BWA+GATK, VCFs were derived from simulated and real sequencing reads. We observed that the prediction models learned by RFR outperformed other algorithms in both simulated and real data. The quality scores of variant calls were highly predictable from informative features of GATK Annotation Modules in the simulated human genome VCF data (R2: 96.7%, 94.4%, and 89.8% for RFR, MLR, and NNR, respectively). The robustness of the proposed data-driven models was consistently maintained in the real human genome VCF data (R2: 97.8% and 96.5% for RFR and MLR, respectively).
下一代测序技术能够实现大规模并行处理,成本低于其他测序技术。在随后的 NGS 数据分析中,主要关注的问题之一是变异调用的可靠性。虽然研究人员可以利用变异调用的原始质量分数,但他们被迫在没有任何质量分数预评估的情况下开始进一步分析。
我们提出了一种机器学习方法,用于估计 BWA+GATK 衍生的变异调用的质量分数。我们分析了质量分数与这些注释之间的相关性,指定了有用的注释作为预测变异质量分数的特征。为了测试预测模型,我们模拟了 24 对 Illumina 测序的双端测序reads,每个 read 有 30x 的覆盖碱基。同时,我们还从序列读取档案中获取了 24 个人类基因组测序 reads,这些 reads 来自 Illumina 双端测序,覆盖率至少为 30x。
使用 BWA+GATK,从模拟和真实测序 reads 中得出了 VCF。我们观察到,RFR 学习的预测模型在模拟和真实数据中均优于其他算法。在模拟的人类基因组 VCF 数据中,从 GATK Annotation Modules 的信息特征中可以高度预测变异调用的质量分数(R2:分别为 96.7%、94.4%和 89.8%,适用于 RFR、MLR 和 NNR)。在真实的人类基因组 VCF 数据中,所提出的数据驱动模型的稳健性得到了一致的保持(R2:分别为 97.8%和 96.5%,适用于 RFR 和 MLR)。