Faculty of Pharmacy, Medical University of Warsaw, Chair and Department of Physical Pharmacy and Bioanalysis, Department of Physical Chemistry, Banacha 1 str., 02-093 Warsaw, Poland.
Molecules. 2020 Mar 30;25(7):1584. doi: 10.3390/molecules25071584.
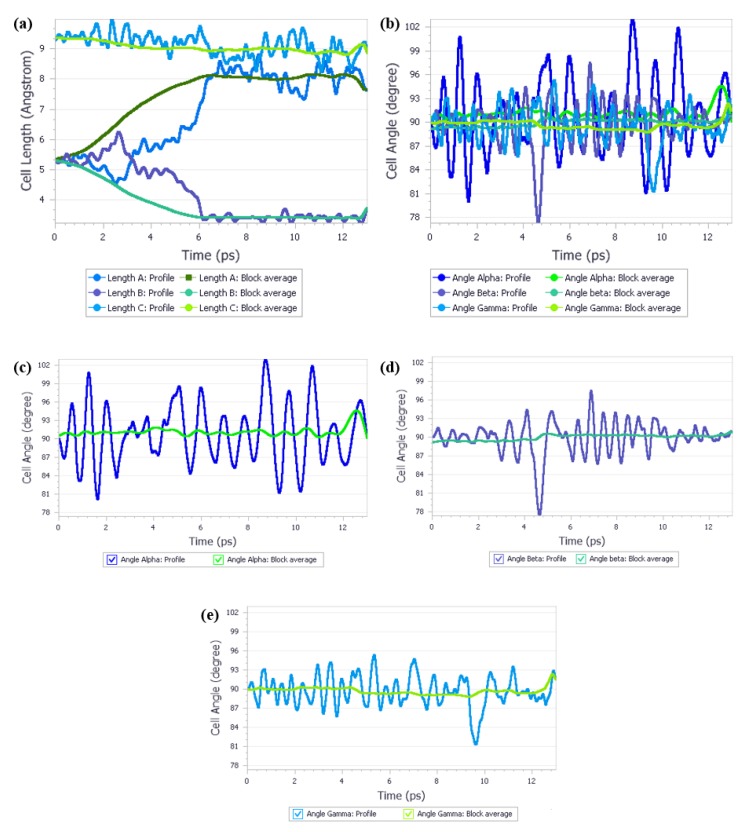
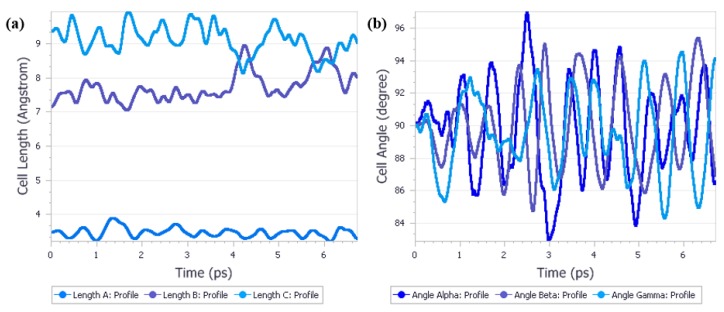
Crystalline urea undergoes polymorphic phase transition induced by high pressure. Form I, which is the most stable form at normal conditions and Form IV, which is the most stable form at 3.10 GPa, not only crystallize in various crystal systems but also differ significantly in the unit cell dimensions. The aim of this study was to determine if it is possible to predict polymorphic phase transitions by optimizing Form I at high pressure and Form IV at low pressure. To achieve this aim, a large number of periodic density functional theory (DFT) calculations were performed using CASTEP. After geometry optimization of Form IV at 0 GPa Form I was obtained, performing energy minimization of Form I at high pressure did not result in Form IV. However, employing quantum molecular isothermal-isobaric (NPT) dynamics calculations enabled to accurately predict this high-pressure transformation. This study shows the potential of different approaches in predicting the polymorphic phase transition and points to the key factors that are necessary to achieve the success.
尿素晶体在高压下经历多晶型相变。在常压下最稳定的形式 I 和在 3.10 GPa 下最稳定的形式 IV,不仅在不同的晶体系统中结晶,而且在单位晶胞尺寸上也有显著差异。本研究的目的是确定是否可以通过在高压下优化形式 I 和在低压下优化形式 IV 来预测多晶型相转变。为了实现这一目标,使用 CASTEP 进行了大量的周期性密度泛函理论(DFT)计算。在 0 GPa 下对形式 IV 进行几何优化后,得到了形式 I,在高压下对形式 I 进行能量最小化并没有得到形式 IV。然而,采用量子分子等温等压(NPT)动力学计算可以准确地预测这种高压转变。本研究展示了不同方法在预测多晶型相转变方面的潜力,并指出了实现成功所需的关键因素。