Alsos Inger Greve, Lavergne Sebastien, Merkel Marie Kristine Føreid, Boleda Marti, Lammers Youri, Alberti Adriana, Pouchon Charles, Denoeud France, Pitelkova Iva, Pușcaș Mihai, Roquet Cristina, Hurdu Bogdan-Iuliu, Thuiller Wilfried, Zimmermann Niklaus E, Hollingsworth Peter M, Coissac Eric
Tromsø Museum, UiT-The Arctic University of Norway, N-9037 Tromsø, Norway.
LECA, Univ. Grenoble Alpes, Univ. Savoie Mont Blanc, CNRS, F-38000 Grenoble, France.
Plants (Basel). 2020 Apr 1;9(4):432. doi: 10.3390/plants9040432.
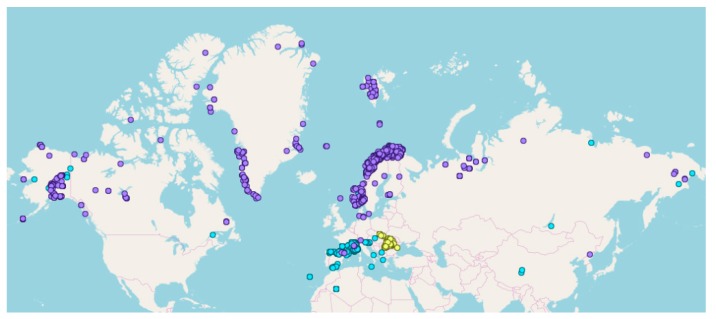
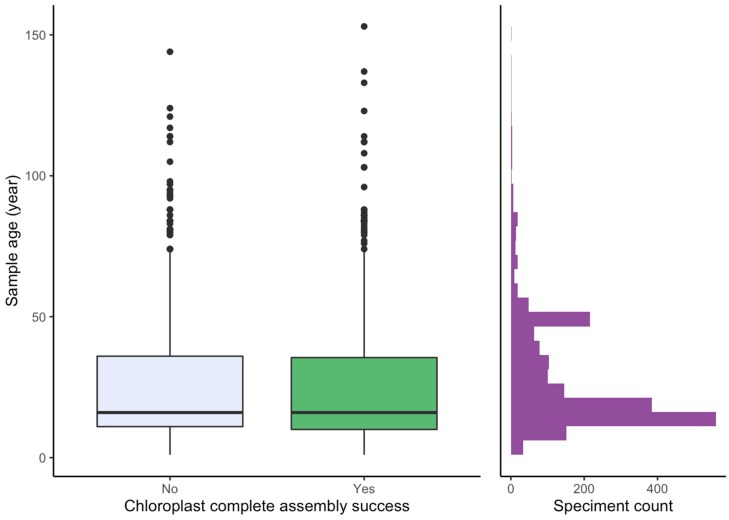
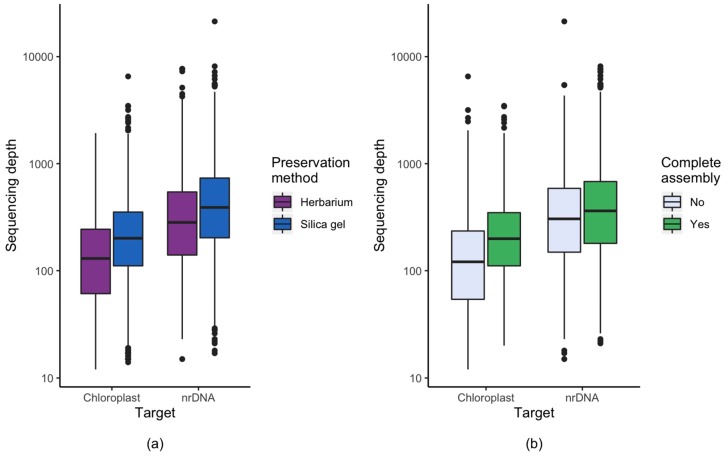
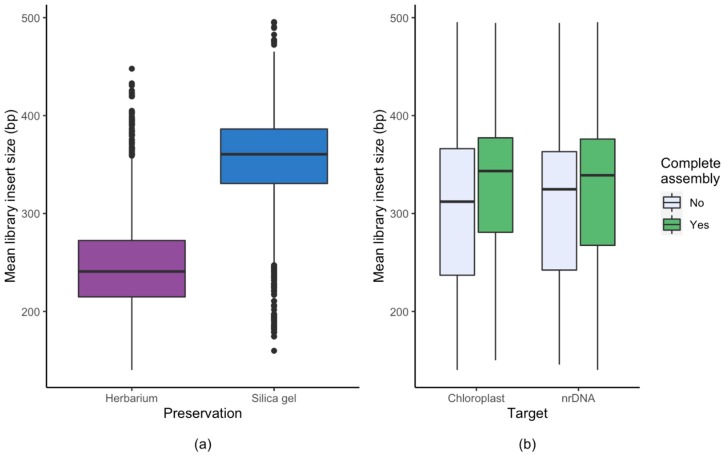
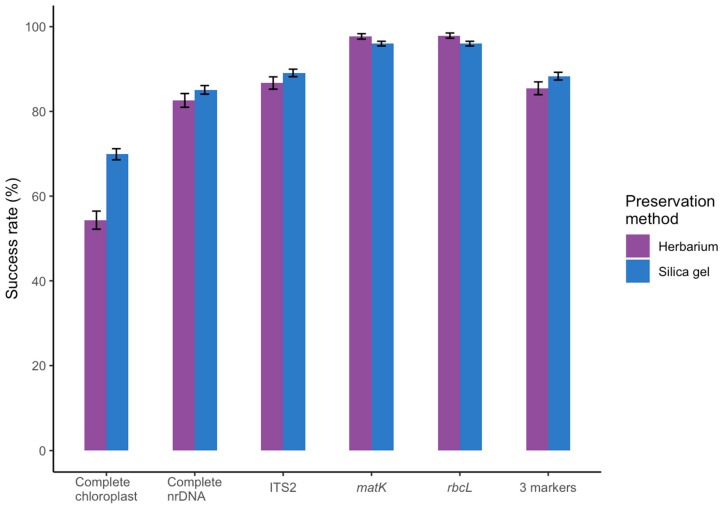
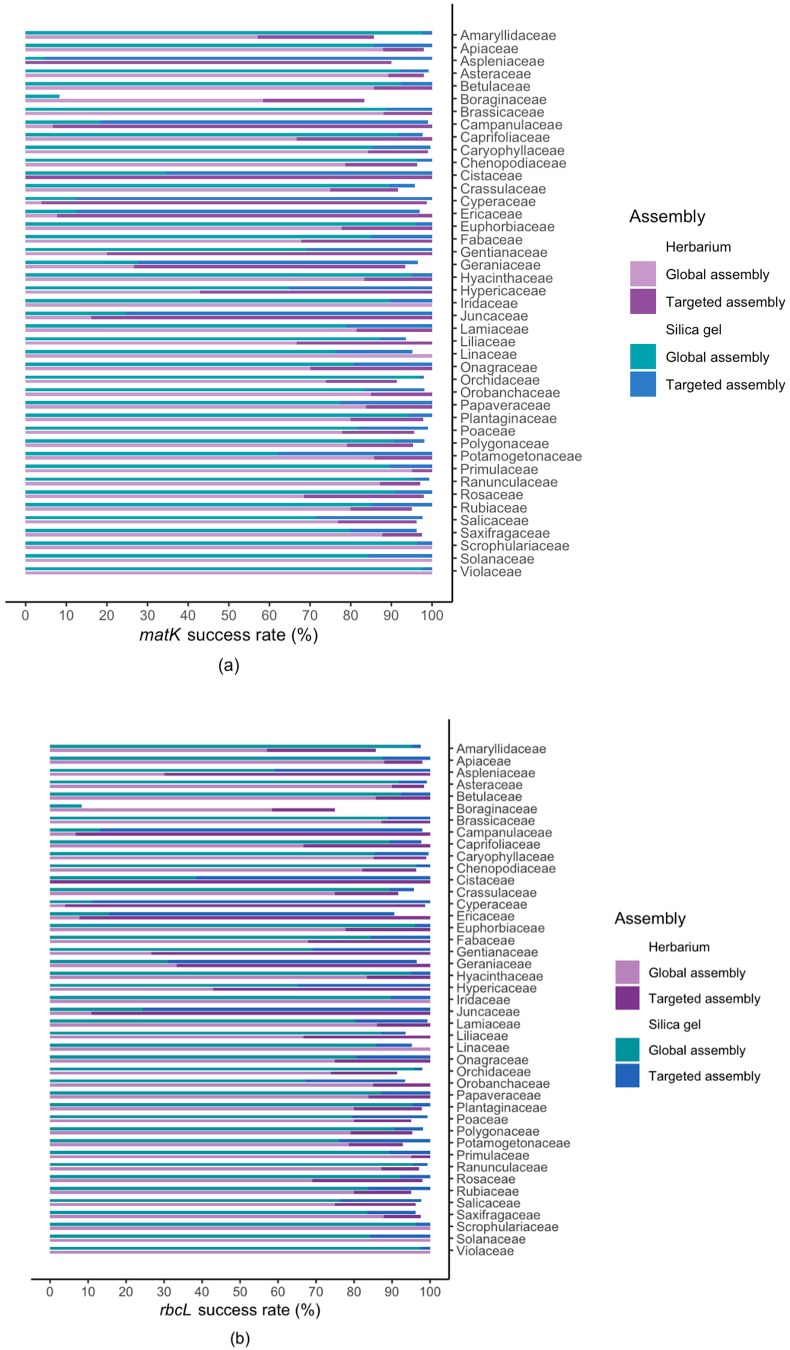
Genome skimming has the potential for generating large data sets for DNA barcoding and wider biodiversity genomic studies, particularly via the assembly and annotation of full chloroplast (cpDNA) and nuclear ribosomal DNA (nrDNA) sequences. We compare the success of genome skims of 2051 herbarium specimens from Norway/Polar regions with 4604 freshly collected, silica gel dried specimens mainly from the European Alps and the Carpathians. Overall, we were able to assemble the full chloroplast genome for 67% of the samples and the full nrDNA cluster for 86%. Average insert length, cover and full cpDNA and rDNA assembly were considerably higher for silica gel dried than herbarium-preserved material. However, complete plastid genomes were still assembled for 54% of herbarium samples compared to 70% of silica dried samples. Moreover, there was comparable recovery of coding genes from both tissue sources (121 for silica gel dried and 118 for herbarium material) and only minor differences in assembly success of standard barcodes between silica dried (89% ITS2, 96% and ) and herbarium material (87% ITS2, 98% and ). The success rate was > 90% for all three markers in 1034 of 1036 genera in 160 families, and only Boraginaceae worked poorly, with 7 genera failing. Our study shows that large-scale genome skims are feasible and work well across most of the land plant families and genera we tested, independently of material type. It is therefore an efficient method for increasing the availability of plant biodiversity genomic data to support a multitude of downstream applications.
基因组浅层测序有潜力为DNA条形码和更广泛的生物多样性基因组研究生成大量数据集,特别是通过完整叶绿体(cpDNA)和核糖体DNA(nrDNA)序列的组装和注释。我们比较了来自挪威/极地地区的2051份植物标本馆标本与主要来自欧洲阿尔卑斯山和喀尔巴阡山脉的4604份新鲜采集、硅胶干燥标本的基因组浅层测序成功率。总体而言,我们能够为67%的样本组装完整的叶绿体基因组,为86%的样本组装完整的nrDNA簇。硅胶干燥样本的平均插入片段长度、覆盖度以及完整的cpDNA和rDNA组装均显著高于植物标本馆保存的材料。然而,与70%的硅胶干燥样本相比,仍有54%的植物标本馆样本组装出了完整的质体基因组。此外,两种组织来源的编码基因回收率相当(硅胶干燥样本为121个,植物标本馆材料为118个),硅胶干燥样本(ITS2为89%,[此处原文缺失部分内容]为96%)和植物标本馆材料(ITS2为87%,[此处原文缺失部分内容]为98%)之间标准条形码的组装成功率仅有微小差异。在160个科的1036个属中,有1034个属的所有三个标记成功率均>90%,只有紫草科效果不佳,有7个属失败。我们的研究表明,大规模基因组浅层测序是可行的,并且在我们测试的大多数陆地植物科和属中都能很好地发挥作用,与材料类型无关。因此,它是一种提高植物生物多样性基因组数据可用性以支持众多下游应用的有效方法。