Lester and Sue Smith Breast Center, Baylor College of Medicine, Houston, TX, 77030, USA.
Department of Molecular and Human Genetics, Baylor College of Medicine, Houston, TX, 77030, USA.
Nat Commun. 2020 Apr 9;11(1):1759. doi: 10.1038/s41467-020-15456-w.
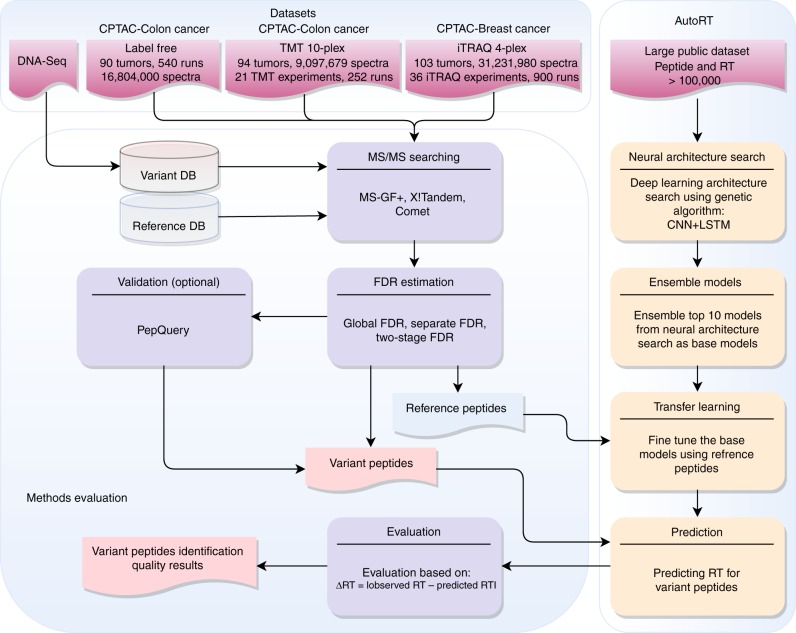
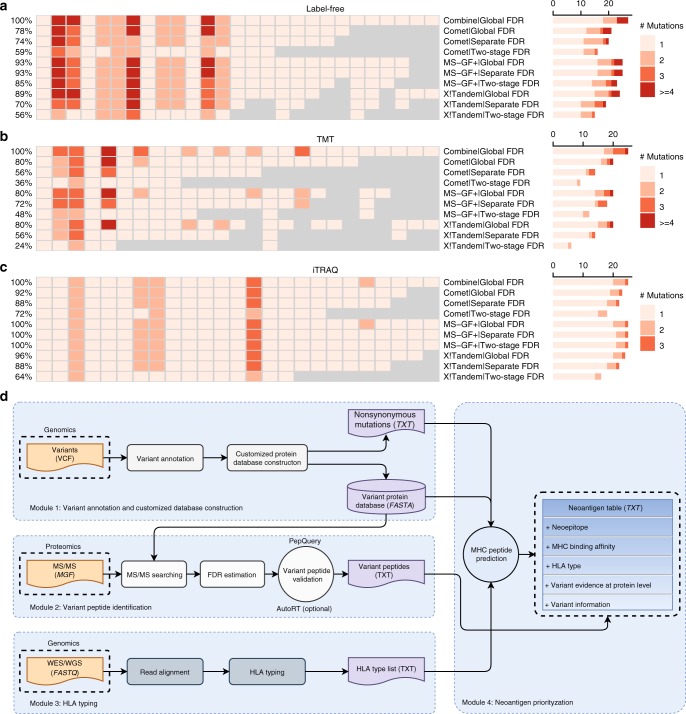
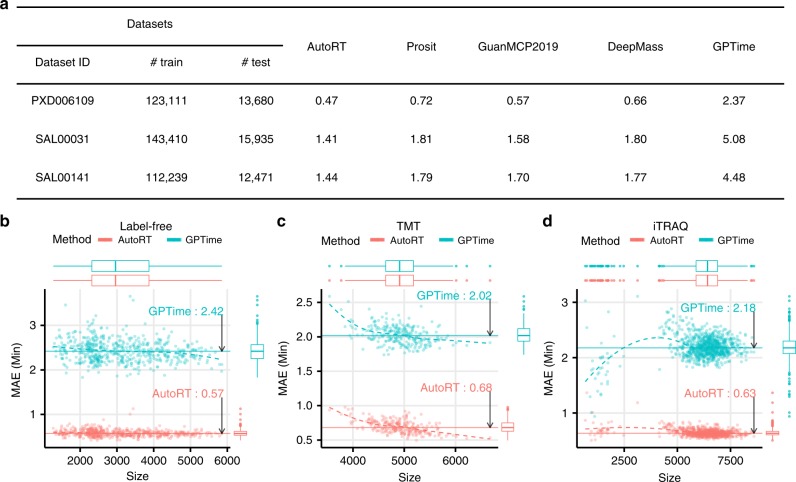
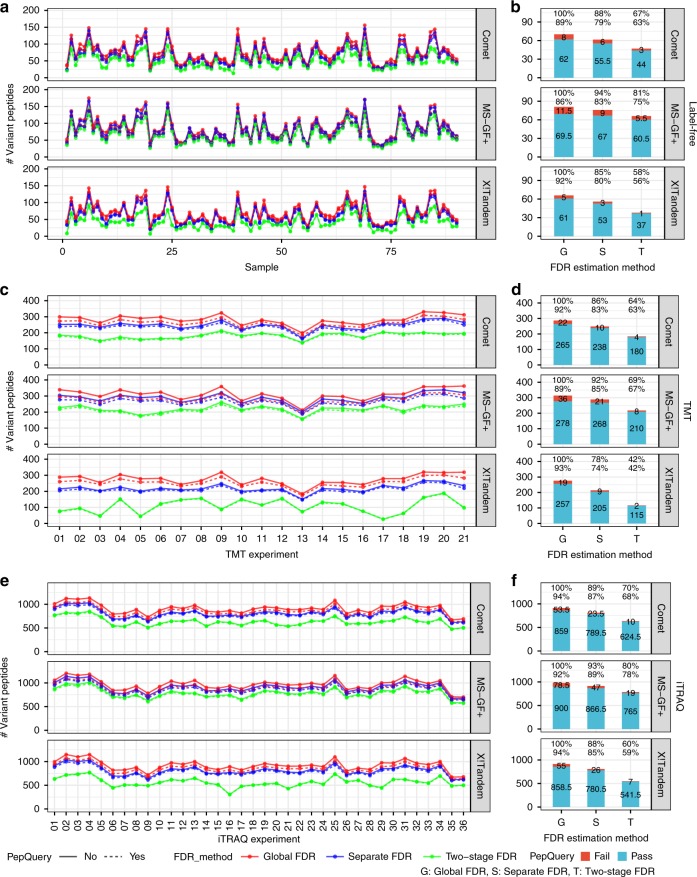
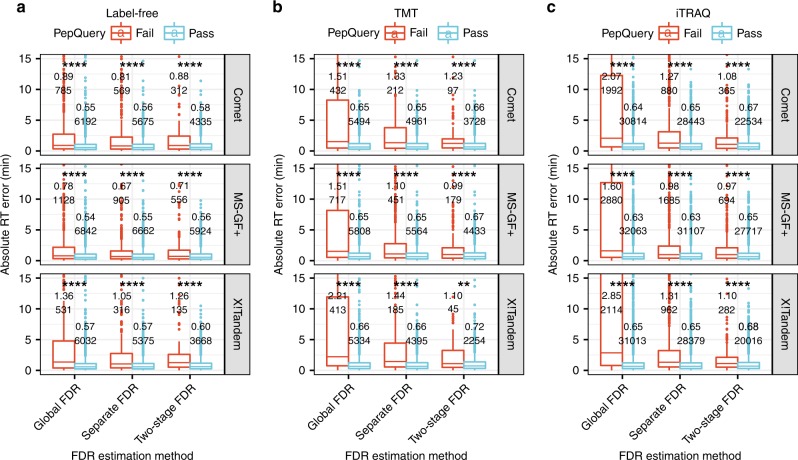
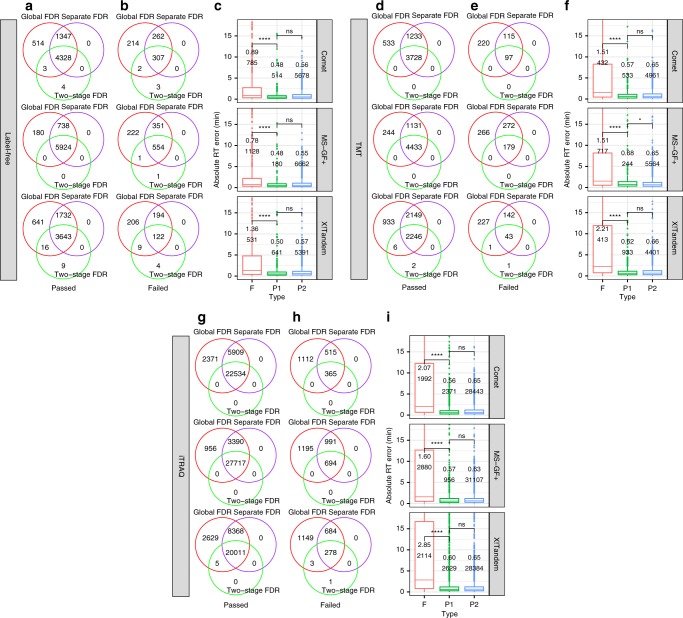
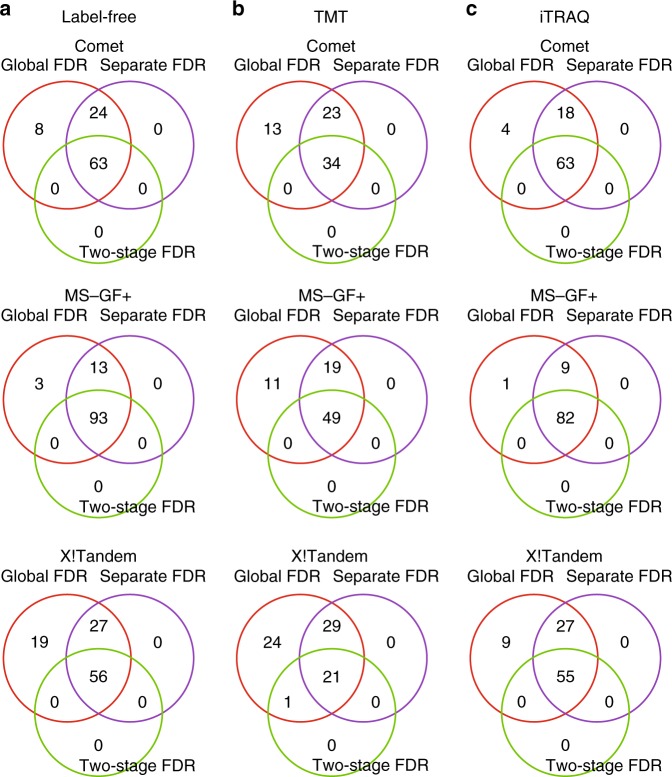
Genomics-based neoantigen discovery can be enhanced by proteomic evidence, but there remains a lack of consensus on the performance of different quality control methods for variant peptide identification in proteogenomics. We propose to use the difference between accurately predicted and observed retention times for each peptide as a metric to evaluate different quality control methods. To this end, we develop AutoRT, a deep learning algorithm with high accuracy in retention time prediction. Analysis of three cancer data sets with a total of 287 tumor samples using different quality control strategies results in substantially different numbers of identified variant peptides and putative neoantigens. Our systematic evaluation, using the proposed retention time metric, provides insights and practical guidance on the selection of quality control strategies. We implement the recommended strategy in a computational workflow named NeoFlow to support proteogenomics-based neoantigen prioritization, enabling more sensitive discovery of putative neoantigens.
基于基因组学的新抗原发现可以通过蛋白质组学证据得到增强,但在蛋白质基因组学中用于鉴定变异肽的不同质量控制方法的性能仍然缺乏共识。我们建议使用每个肽的准确预测和观察到的保留时间之间的差异作为评估不同质量控制方法的指标。为此,我们开发了 AutoRT,这是一种在保留时间预测方面具有高精度的深度学习算法。使用不同的质量控制策略对三个癌症数据集(共 287 个肿瘤样本)进行分析,得到的鉴定出的变异肽和潜在新抗原的数量有很大差异。我们使用建议的保留时间指标进行的系统评估,为质量控制策略的选择提供了深入的见解和实用指导。我们在名为 NeoFlow 的计算工作流程中实现了推荐的策略,以支持基于蛋白质基因组学的新抗原优先级排序,从而更敏感地发现潜在的新抗原。