Westermayr Julia, Gastegger Michael, Marquetand Philipp
Institute of Theoretical Chemistry, Faculty of Chemistry, University of Vienna, Währinger Str. 17, 1090 Vienna, Austria.
Machine Learning Group, Technical University of Berlin, 10587 Berlin, Germany.
J Phys Chem Lett. 2020 May 21;11(10):3828-3834. doi: 10.1021/acs.jpclett.0c00527. Epub 2020 May 1.
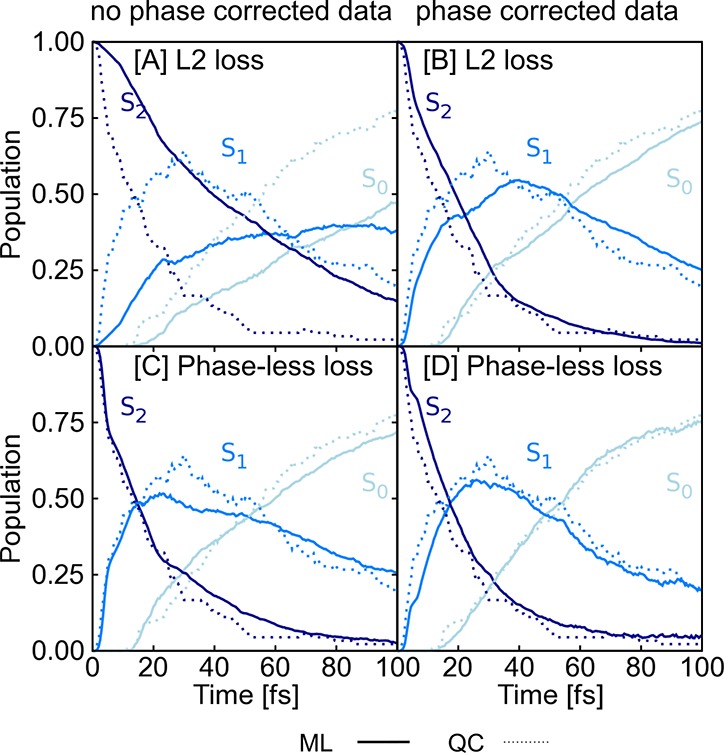
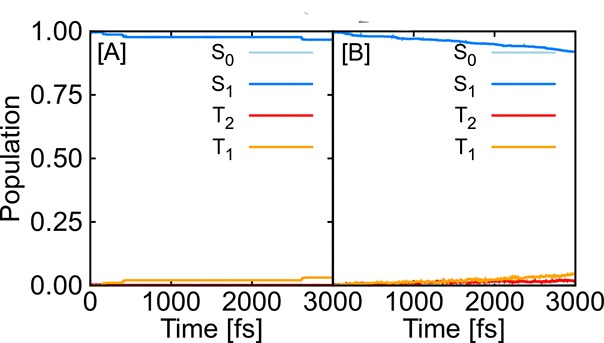
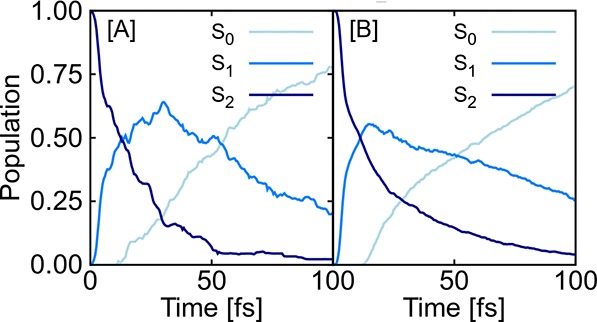
In recent years, deep learning has become a part of our everyday life and is revolutionizing quantum chemistry as well. In this work, we show how deep learning can be used to advance the research field of photochemistry by learning all important properties-multiple energies, forces, and different couplings-for photodynamics simulations. We simplify such simulations substantially by (i) a phase-free training skipping costly preprocessing of raw quantum chemistry data; (ii) rotationally covariant nonadiabatic couplings, which can either be trained or (iii) alternatively be approximated from only ML potentials, their gradients, and Hessians; and (iv) incorporating spin-orbit couplings. As the deep-learning method, we employ SchNet with its automatically determined representation of molecular structures and extend it for multiple electronic states. In combination with the molecular dynamics program SHARC, our approach termed SchNarc is tested on two polyatomic molecules and paves the way toward efficient photodynamics simulations of complex systems.
近年来,深度学习已成为我们日常生活的一部分,同时也正在给量子化学带来变革。在这项工作中,我们展示了如何通过学习光动力学模拟的所有重要属性(多种能量、力和不同耦合),利用深度学习推动光化学研究领域的发展。我们通过以下方式大幅简化此类模拟:(i)无相位训练,跳过原始量子化学数据的昂贵预处理;(ii)旋转协变非绝热耦合,其既可以进行训练,或者(iii)也可以仅从机器学习势、其梯度和海森矩阵进行近似;以及(iv)纳入自旋轨道耦合。作为深度学习方法,我们采用具有自动确定分子结构表示的SchNet,并将其扩展到多个电子态。结合分子动力学程序SHARC,我们称为SchNarc的方法在两个多原子分子上进行了测试,为复杂系统的高效光动力学模拟铺平了道路。