Hamilton Glaucoma Center and Shiley Eye Center, The Viterbi Family Department of Ophthalmology, University of California San Diego, La Jolla, CA, USA.
National Center for Microscopy and Imaging Research and Department of Neurosciences, University of California San Diego, La Jolla, CA, USA.
Cell Death Dis. 2020 Apr 20;11(4):254. doi: 10.1038/s41419-020-2456-6.
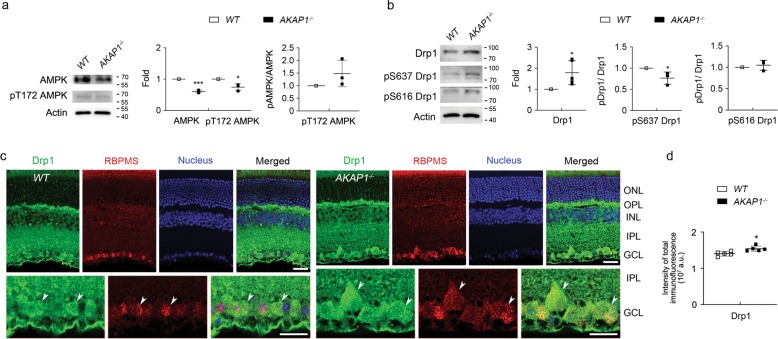
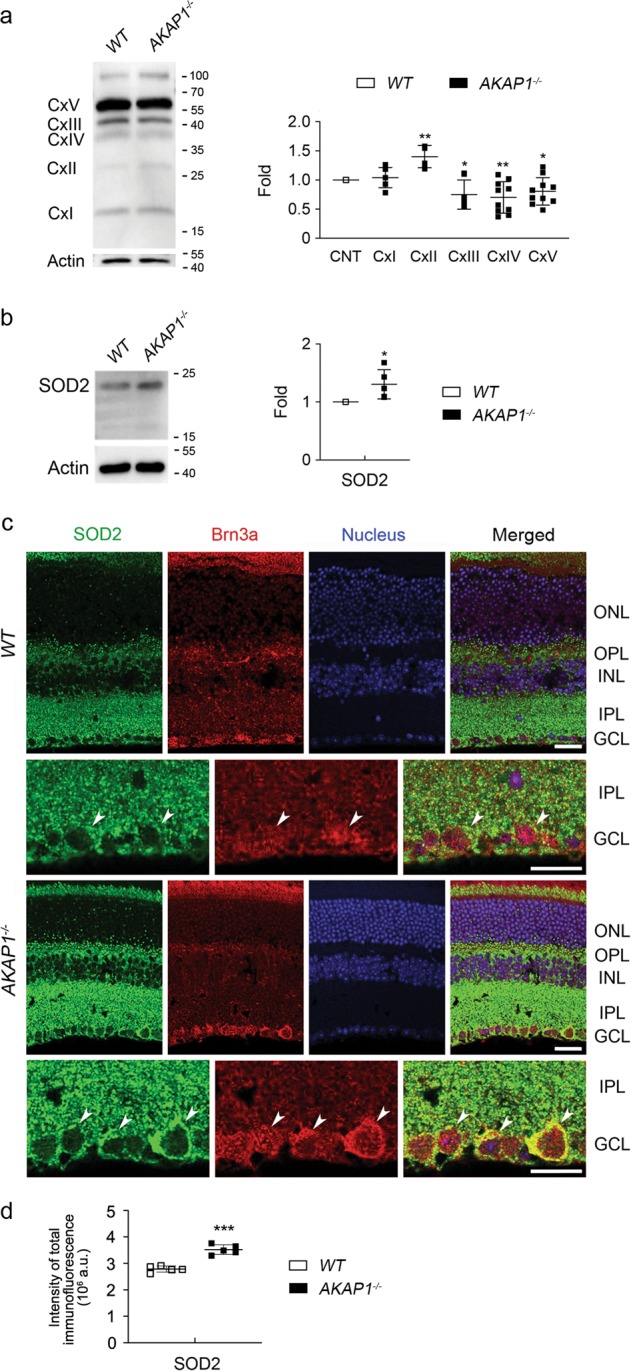
Impairment of mitochondrial structure and function is strongly linked to glaucoma pathogenesis. Despite the widely appreciated disease relevance of mitochondrial dysfunction and loss, the molecular mechanisms underlying mitochondrial fragmentation and metabolic stress in glaucoma are poorly understood. We demonstrate here that glaucomatous retinal ganglion cells (RGCs) show loss of A-kinase anchoring protein 1 (AKAP1), activation of calcineurin (CaN) and reduction of dynamin-related protein 1 (Drp1) phosphorylation at serine 637 (Ser637). These findings suggest that AKAP1-mediated phosphorylation of Drp1 at Ser637 has a critical role in RGC survival in glaucomatous neurodegeneration. Male mice lacking AKAP1 show increases in CaN and total Drp1 levels, as well as a decrease in Drp1 phosphorylation at Ser637 in the retina. Ultrastructural analysis of mitochondria shows that loss of AKAP1 triggers mitochondrial fragmentation and loss, as well as mitophagosome formation in RGCs. Loss of AKAP1 deregulates oxidative phosphorylation (OXPHOS) complexes (Cxs) by increasing CxII and decreasing CxIII-V, leading to metabolic and oxidative stress. Also, loss of AKAP1 decreases Akt phosphorylation at Serine 473 (Ser473) and threonine 308 (Thr308) and activates the Bim/Bax signaling pathway in the retina. These results suggest that loss of AKAP1 has a critical role in RGC dysfunction by decreasing Drp1 phosphorylation at Ser637, deregulating OXPHOS, decreasing Akt phosphorylation at Ser473 and Thr308, and activating the Bim/Bax pathway in glaucomatous neurodegeneration. Thus, we propose that overexpression of AKAP1 or modulation of Drp1 phosphorylation at Ser637 are potential therapeutic strategies for neuroprotective intervention in glaucoma and other mitochondria-related optic neuropathies.
线粒体结构和功能的损伤与青光眼的发病机制密切相关。尽管人们广泛认识到线粒体功能障碍和丧失与疾病的相关性,但青光眼中线粒体碎片化和代谢应激的分子机制仍知之甚少。我们在这里证明,青光眼性视网膜神经节细胞 (RGC) 表现出 A-激酶锚定蛋白 1 (AKAP1) 的丧失、钙调神经磷酸酶 (CaN) 的激活和动力相关蛋白 1 (Drp1) 丝氨酸 637 (Ser637) 磷酸化的减少。这些发现表明,AKAP1 介导的 Drp1 在 Ser637 处的磷酸化在青光眼神经退行性变中 RGC 存活中具有关键作用。缺乏 AKAP1 的雄性小鼠在视网膜中表现出 CaN 和总 Drp1 水平增加,以及 Drp1 在 Ser637 处磷酸化减少。线粒体的超微结构分析表明,AKAP1 的缺失会触发 RGC 中线粒体的碎片化和丧失,以及噬线粒体的形成。AKAP1 的缺失通过增加 CxII 和减少 CxIII-V 来扰乱氧化磷酸化 (OXPHOS) 复合物 (Cxs),导致代谢和氧化应激。此外,AKAP1 的缺失减少了 Akt 在丝氨酸 473 (Ser473) 和苏氨酸 308 (Thr308) 处的磷酸化,并激活了视网膜中的 Bim/Bax 信号通路。这些结果表明,AKAP1 的缺失通过降低 Drp1 在 Ser637 处的磷酸化、扰乱 OXPHOS、减少 Akt 在 Ser473 和 Thr308 处的磷酸化以及激活 Bim/Bax 通路,在青光眼性神经退行性变中对 RGC 功能障碍具有关键作用。因此,我们提出 AKAP1 的过表达或 Drp1 在 Ser637 处磷酸化的调节可能是青光眼和其他与线粒体相关的视神经病变中神经保护干预的潜在治疗策略。