Chen Ya, Mathai Neann, Kirchmair Johannes
Center for Bioinformatics (ZBH), Department of Computer Science, Faculty of Mathematics, Informatics and Natural Sciences, Universität Hamburg, 20146 Hamburg, Germany.
Department of Chemistry and Computational Biology Unit (CBU), University of Bergen, N-5020 Bergen, Norway.
J Chem Inf Model. 2020 Jun 22;60(6):2858-2875. doi: 10.1021/acs.jcim.0c00161. Epub 2020 May 5.
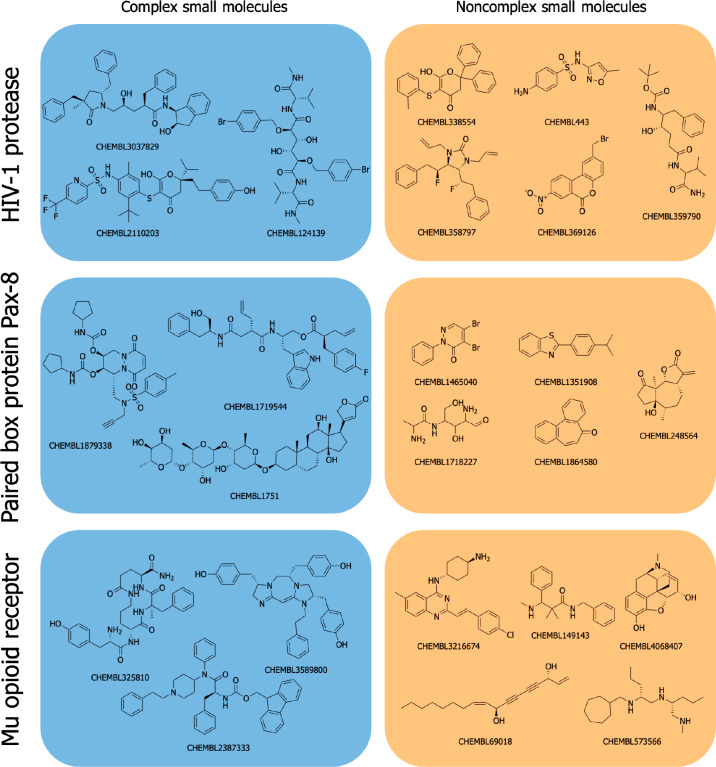
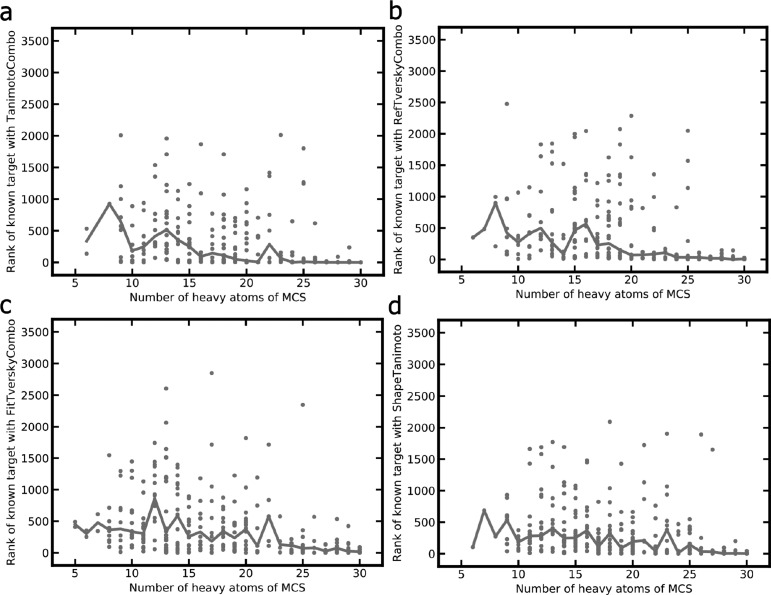
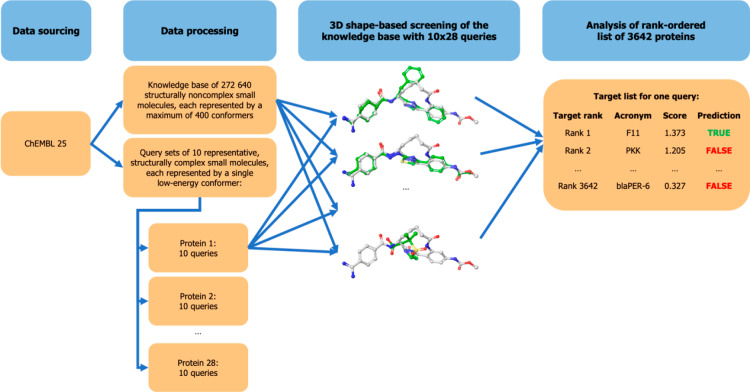
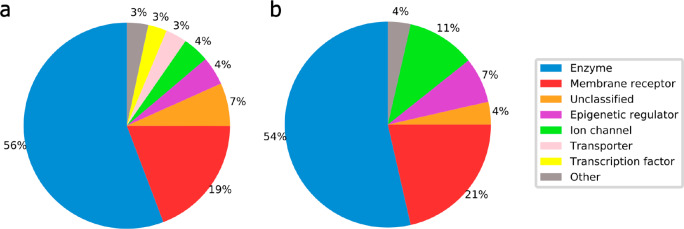
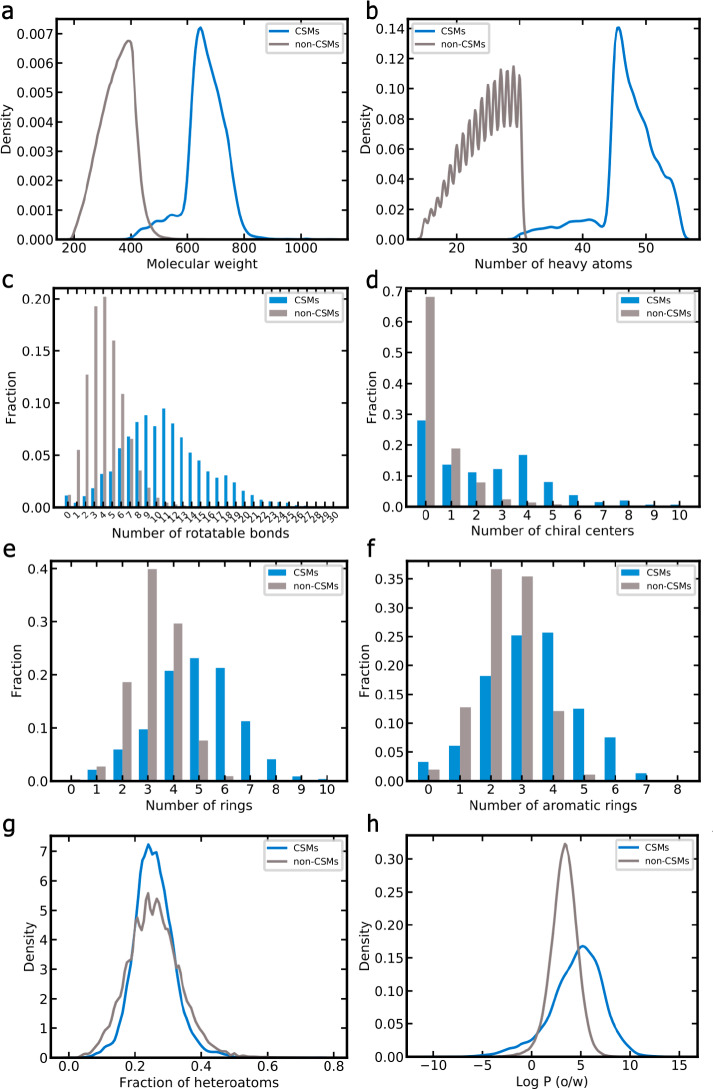
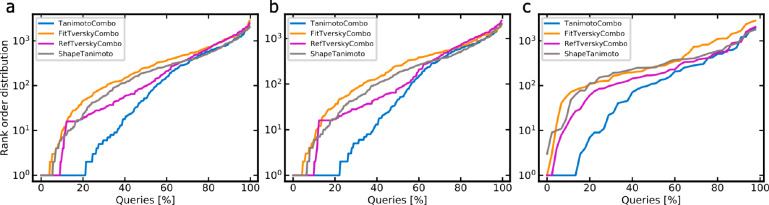
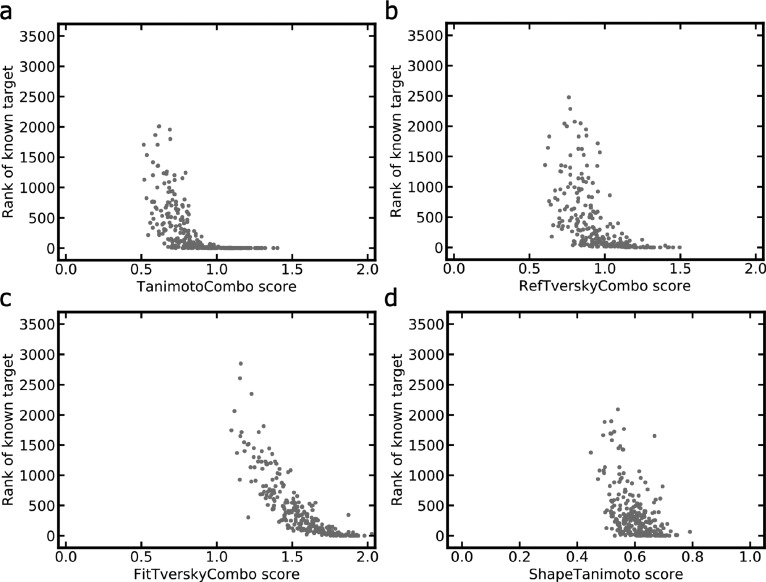
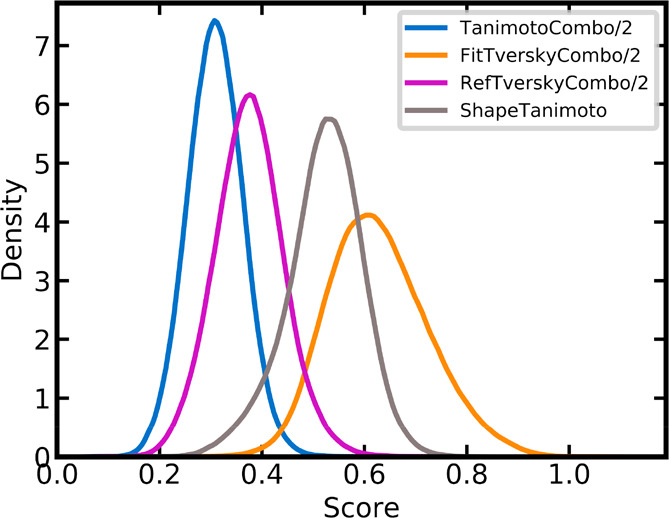
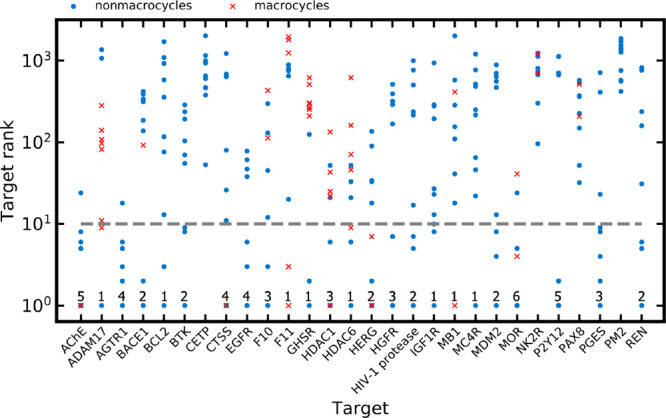
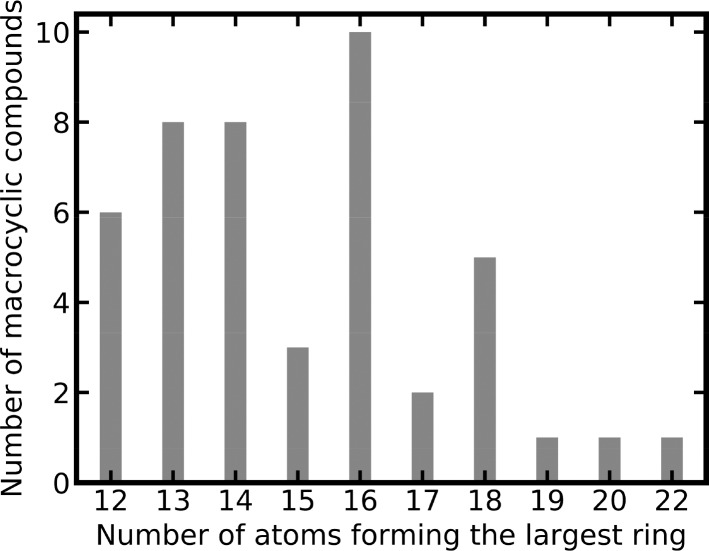
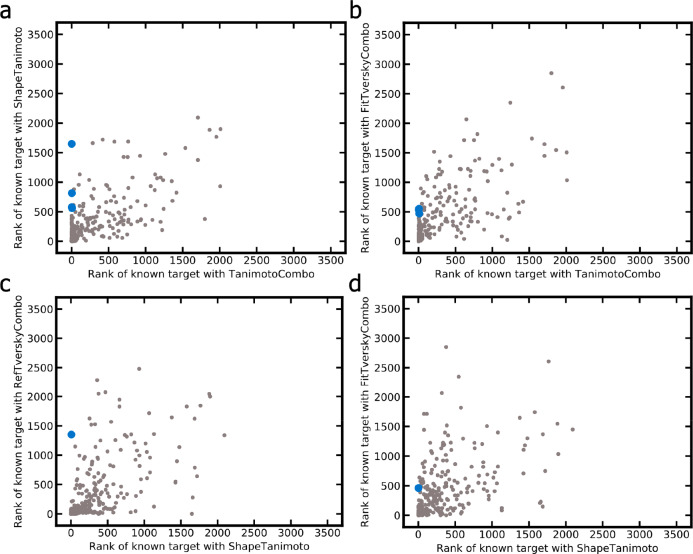
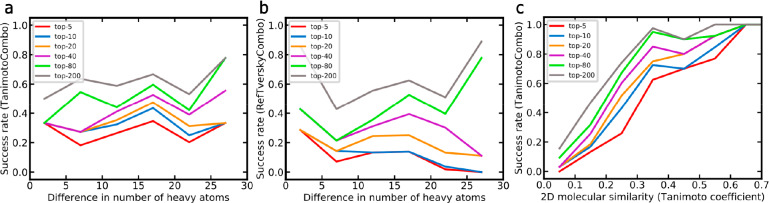
A plethora of similarity-based, network-based, machine learning, docking and hybrid approaches for predicting the macromolecular targets of small molecules are available today and recognized as valuable tools for providing guidance in early drug discovery. With the increasing maturity of target prediction methods, researchers have started to explore ways to expand their scope to more challenging molecules such as structurally complex natural products and macrocyclic small molecules. In this work, we systematically explore the capacity of an alignment-based approach to identify the targets of structurally complex small molecules (including large and flexible natural products and macrocyclic compounds) based on the similarity of their 3D molecular shape to noncomplex molecules (i.e., more conventional, "drug-like", synthetic compounds). For this analysis, query sets of 10 representative, structurally complex molecules were compiled for each of the 28 pharmaceutically relevant proteins. Subsequently, ROCS, a leading shape-based screening engine, was utilized to generate rank-ordered lists of the potential targets of the 28 × 10 queries according to the similarity of their 3D molecular shapes with those of compounds from a knowledge base of 272 640 noncomplex small molecules active on a total of 3642 different proteins. Four of the scores implemented in ROCS were explored for target ranking, with the TanimotoCombo score consistently outperforming all others. The score successfully recovered the targets of 30% and 41% of the 280 queries among the top-5 and top-20 positions, respectively. For 24 out of the 28 investigated targets (86%), the method correctly assigned the first rank (out of 3642) to the target of interest for at least one of the 10 queries. The shape-based target prediction approach showed remarkable robustness, with good success rates obtained even for compounds that are clearly distinct from any of the ligands present in the knowledge base. However, complex natural products and macrocyclic compounds proved to be challenging even with this approach, although cases of complete failure were recorded only for a small number of targets.
如今,有大量基于相似性、基于网络、机器学习、对接和混合的方法可用于预测小分子的大分子靶点,并且这些方法被认为是在早期药物发现中提供指导的有价值工具。随着靶点预测方法日益成熟,研究人员已开始探索将其范围扩展到更具挑战性的分子的方法,例如结构复杂的天然产物和大环小分子。在这项工作中,我们系统地探索了一种基于比对的方法的能力,该方法基于结构复杂的小分子(包括大的和柔性的天然产物以及大环化合物)与非复杂分子(即更传统的“类药物”合成化合物)的三维分子形状相似性来识别其靶点。为了进行此分析,针对28种与药物相关的蛋白质中的每一种,编制了10个具有代表性的结构复杂分子的查询集。随后,使用领先的基于形状的筛选引擎ROCS,根据28×10个查询的三维分子形状与来自272640个非复杂小分子知识库中化合物的三维分子形状的相似性,生成潜在靶点的排名列表,这些非复杂小分子对总共3642种不同蛋白质具有活性。研究了ROCS中实现的四个分数用于靶点排名,其中TanimotoCombo分数始终优于所有其他分数。该分数分别在排名前5和前20的位置成功找到了280个查询中30%和41%的靶点。对于28个研究靶点中的24个(86%),该方法至少在10个查询中的一个中将感兴趣的靶点正确地排在了第一位(在3642个中)。基于形状的靶点预测方法显示出显著的稳健性,即使对于与知识库中存在的任何配体明显不同的化合物,也能获得良好的成功率。然而,即使采用这种方法,复杂的天然产物和大环化合物仍然具有挑战性,尽管仅记录了少数靶点的完全失败情况。